






一、软件介绍
随着测序技术的快速发展,产生了大量的宏基因组数据,这些不同来源的宏基因组数据会包含很多重复数据,为了对这些数据进行去冗余,查看文献发现了两种主流的去冗余软件,cd-hit 和 dRep。
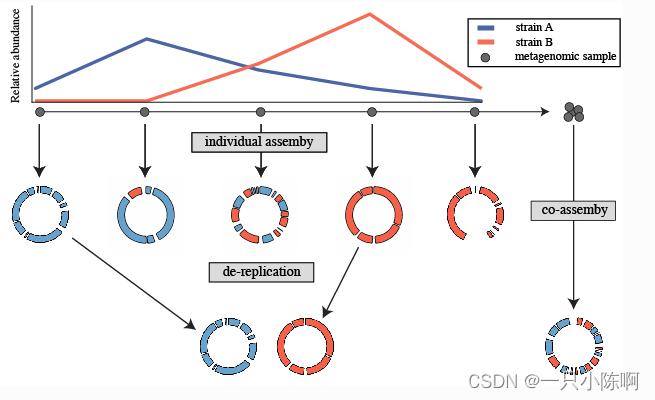
图片引用:(Overview — drep 2.0.0 documentation)
CD-HIT:
- CD-HIT 是一个经典的去冗余工具,广泛用于序列聚类和去冗余。
- 它适用于 DNA/RNA 序列和蛋白序列的去冗余,以及两个数据集之间的比较。
- CD-HIT 的性能在大型基因组集上可能受到限制,尤其是对于草图质量的基因组。
- 它的去冗余效果通常较好,但在处理大规模数据时,计算时间可能较长。
dRep:
- dRep 是由加州大学伯克利分校的 Jillian F Banfield 组开发的,专门用于微生物基因组的快速去冗余。
- 它通过结合快速但不准确的基因组距离估算和较慢但准确的平均核苷酸同一性测量,减少了成对基因组比较的计算时间。
- 相较于先前的算法,dRep 的速度提高了 28 倍,同时保持了完美的查全率和精确度。
- 它适用于从时间序列数据集中恢复基因组,以及从多个样本中识别基本相同的基因组并选择最佳基因组。
二、软件安装
CD-HIT:
使用conda 能够直接安装,比较简单:

dRep :
dRep的安装较为复杂,github中推荐使用pip安装,并且它的安装涉及多个依赖包(本文推荐按照官方安装流程来安装):
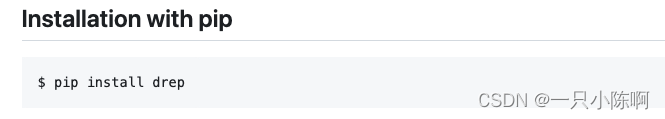

1、首先使用pip 安装 dRep
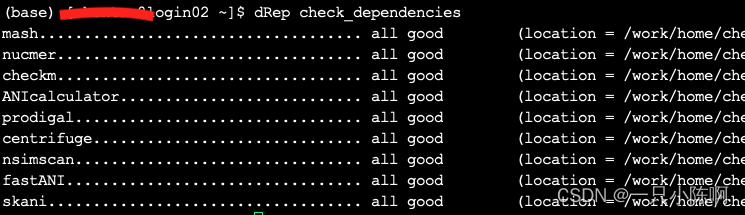
pip install dRep安装完成后,使用命令检测依赖包安装情况(Mash和MUMmer是必须的,其它的若是用不着可以不安装):
dRep check_dependencies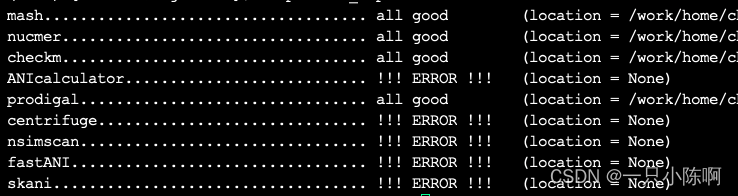
2、根据提示安装依赖,包括使用conda、pip、以及编译安装:
(1)首先安装必须依赖包MUMmer、Mash:
conda install mash mummer(2)成功安装后再安装其它依赖包:
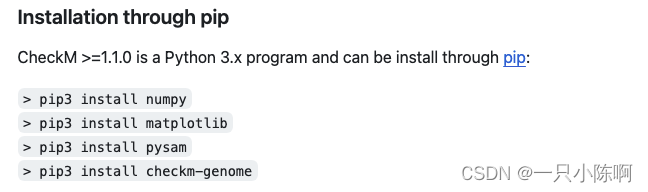
- checkM 是一款利用基因的单拷贝性来有效的评估基因组草图的质量,包括完整度、污染度序列分布等。官网推荐了两种安装途径:

在完成安装之后要下载数据包,不然会使用报错,步骤如下,在软件文件夹构建新文件夹,然后下载数据包,解压,最后指定该文件夹:
mkdir ~/software/CheckM
cd ~/software/CheckM
wget https://data.ace.uq.edu.au/public/CheckM_databases/checkm_data_2015_01_16.tar.gz
tar -zxvf checkm_data_2015_01_16.tar.gz
checkm data setRoot $Path_checkm_data
checkm -h- prodigal、centrifuge、fastANI、skani使用conda安装:
conda install prodigal centrifuge fastani skani- nsimsan 使用编译安装,先从github中下载文件,然后安装:
cd ~/software
wget https://link.zhihu.com/?target=https%3A//github.com/abadona/qsimscan/archive/refs/heads/master.zip
unzip master.zip #(下载后文件名可能会乱码,改下名字或者在MAC、Windows中下载后再传到Linux)
cd qsimscan-master
make #成功后加入环境变量即可
- ANIcalculator 使用编译安装,首先下载对应的安装包(安装包中还有nsimsan,我们已经装好了,没有安装的也可以直接通过这里使用):
cd ~/software
wget https://ani.jgi.doe.gov/download_files/ANIcalculator_v1.tgz
tar -zxvf ANIcalculator_v1.tgz
cd ANIcalculator_v1 #发现该文件夹内包含ANIcalculator和nsimscan两个软件,将该文件夹加入环境变量即可全部安装完成后再次检验依赖包,全部安装成功!
三、使用介绍
CD-HIT:
cd-hit能够支持 .fasta 和 fasta.gz 格式的文件作为输入 ,但是好像不支持多文件输入
#首先将要去冗余的数据合并成一个文件:
cat 1.fasta 2.fasta 3.fasta > Merge.fasta
#使用cd-hit去冗余:
cd-hit-est -i Merge.fasta -o Merge.cdhit.fa -c 0.95 -n 10 -d 0 -M 16000 -T 8dRep :
官方帮助文档(Quick Start — drep 2.0.0 documentation)
dRep是一款支持多文件,多格式的软件,并且能够对数据使用checkM分析输入数据的软件
dRep dereplicate outout_directory -g path/to/genomes/*.fasta两种软件都各自具有优缺点,可以根据自己数据的类型来选择,由于我要分析的数据类型比较多,所以选择了dRep对数据进行去冗余!
感谢阅读!















 1679
1679











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









