







autodock-vina分子对接
1.蛋白质的处理
从蛋白库里获取蛋白质的初始结构文件http://www.rcsb.org/
这里我们选用3HTB蛋白复合物文件,用可视化软件打开pdb文件,这里选用pymol软件查看
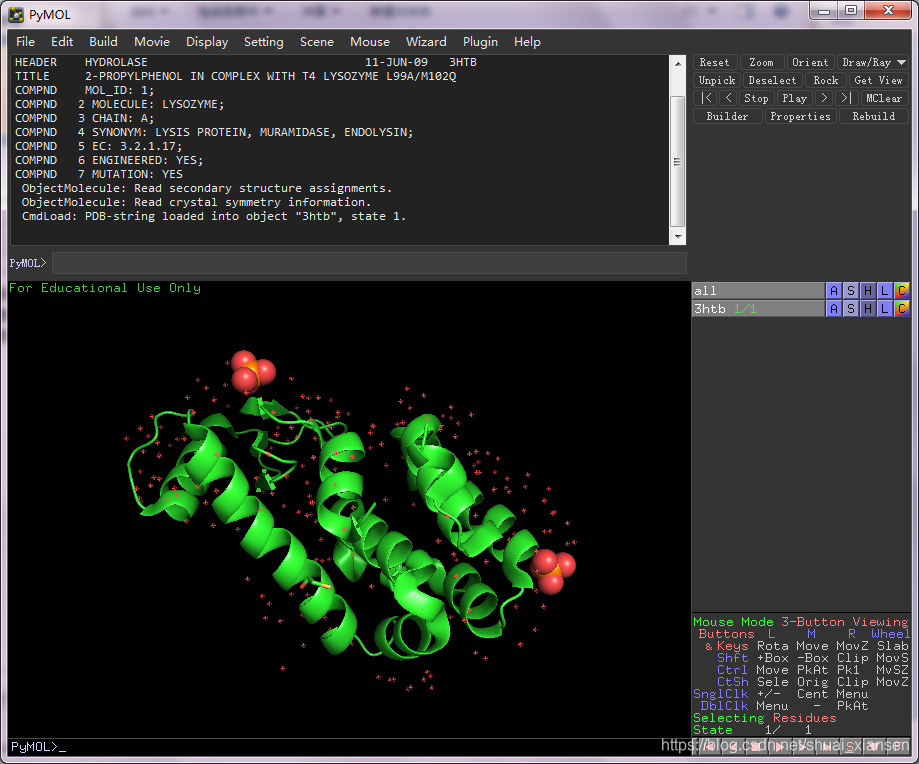
我们去除掉水分子以及配体,在右侧3htb中点A,选择remove waters ,再点中右下角S,拖动序列栏,在序列栏中选中其他小分子配体,在右侧sele中点A,选择remove atoms,删除配体
现在得到了一个处理完的蛋白受体分子,保存为pdb格式,重命名rep.pdb
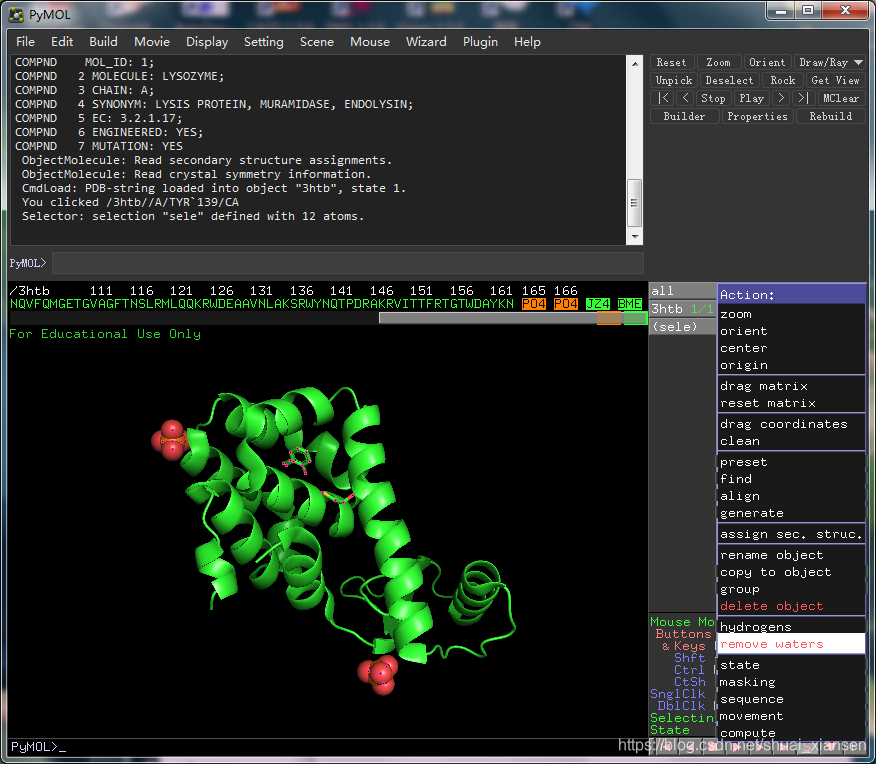
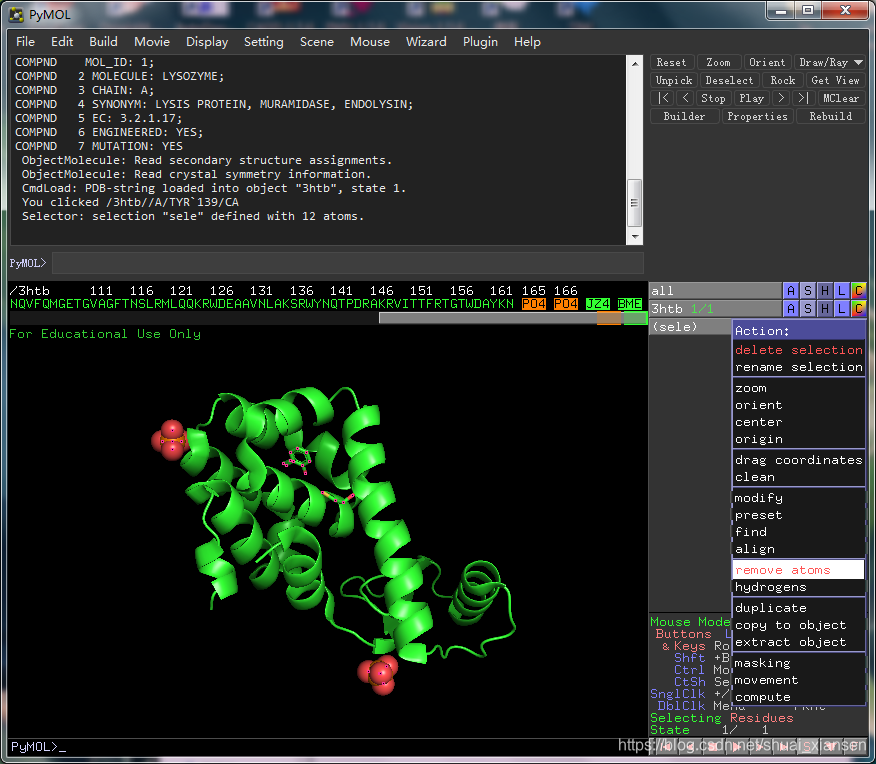
打开rep.pdb查看蛋白是否处理干净
2.配体的处理
小分子配体可以从网上下载也可以自己用Chemdraw画,这里我准备了JZ4的小分子配体
3.用autodock开始对接
将rep.pdb与JZ4.pdb文件导入autodock tools中
用Edit->Hydrogens->Add为受体分子加上H原子,保存修改好的受体分子
使用Ligand下拉菜单中的Torsion Tree一系列工具,设置配体分子的可扭转键,保存配体分子JZ4.pdbqt文件
Grid->Macormolecule->choose 选择rep受体文件,保存rep.pdbqt文件
Grid->set map types ->choose ligand选择JZ4配体文件
Grid Box准备一个大盒子,将大分子的活性位点包括在里面,Grid Options菜单中File->Close Saving current保存。Grid->Output->Save GPF将Grid参数保存成格子参数文件
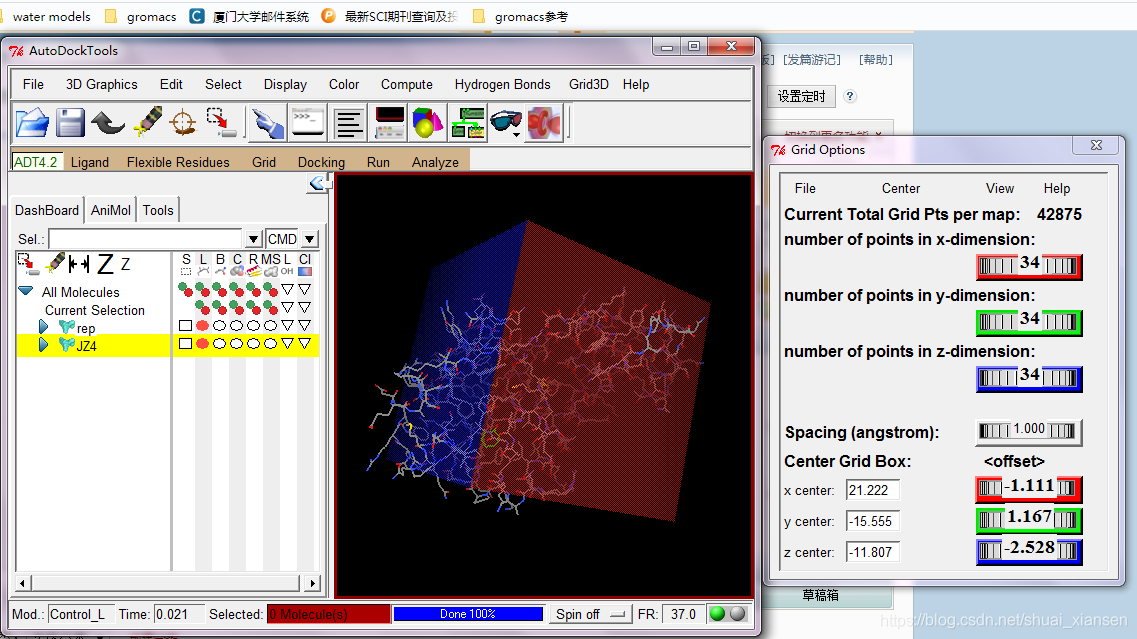
这里我们用vina来进行模拟
vina用cmd命令行运行,脚本为conf.tx文件,内容如下,可增加参数
receptor = rep.pdbqt
ligand=JZ4.pdbqt
center_x = 22.333
center_y = -16.724
center_z = -9.278
size_x = 34
size_y = 34
size_z = 34
energy_range = 5
exhaustiveness = 400
num_modes = 20
运行vina
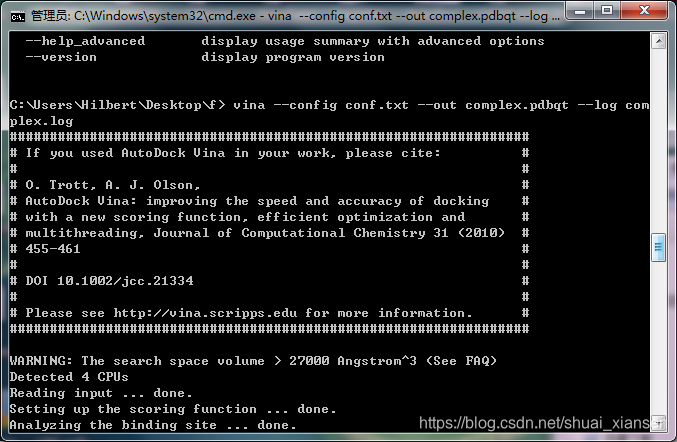
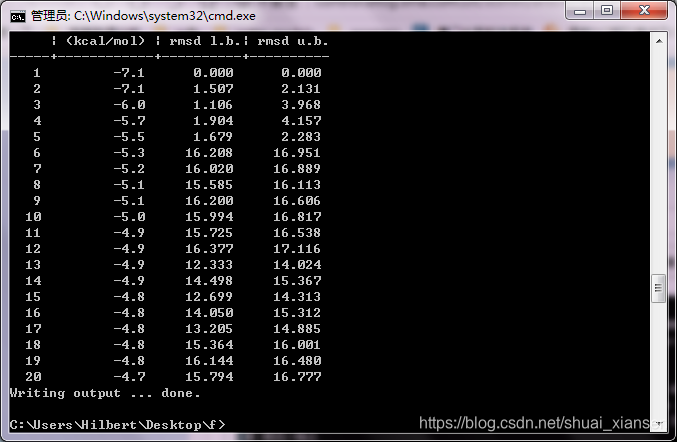
运行结束,生成了19个构象,并分别打分
将生成的complex.pdbqt文件与rep.pdb文件一起导入到pymol中观察
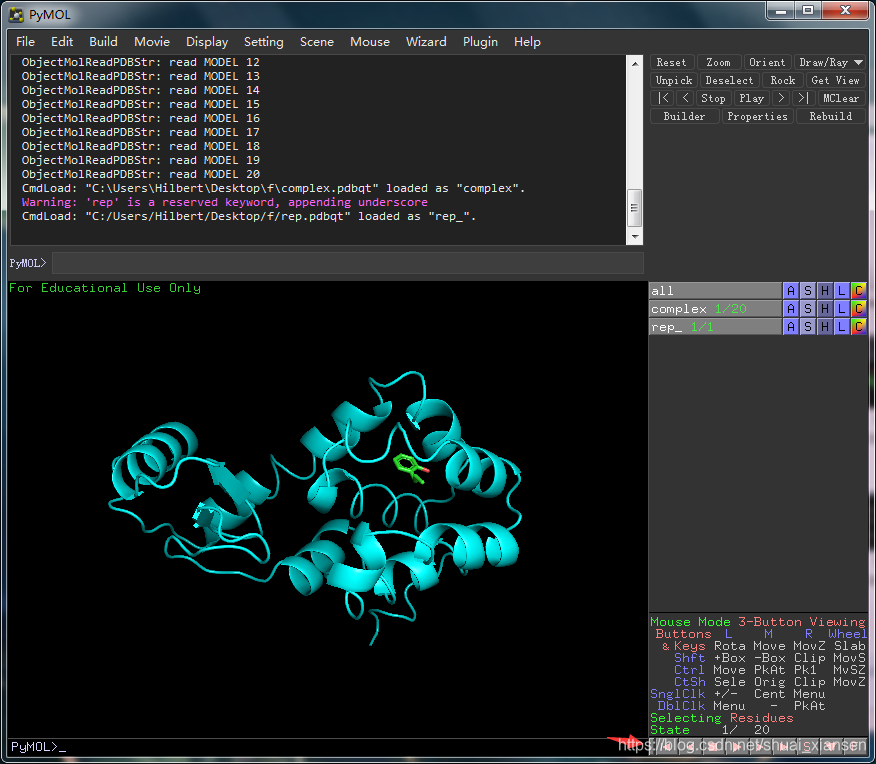
第一张结合位点还可以,结合能打分也不错,一般取第一的构象,然后给配体加氢,导出保存
因为JZ4配体是从复合物里抽取出来的,所以可以将对接后的小分子与原配体做一个叠合对比
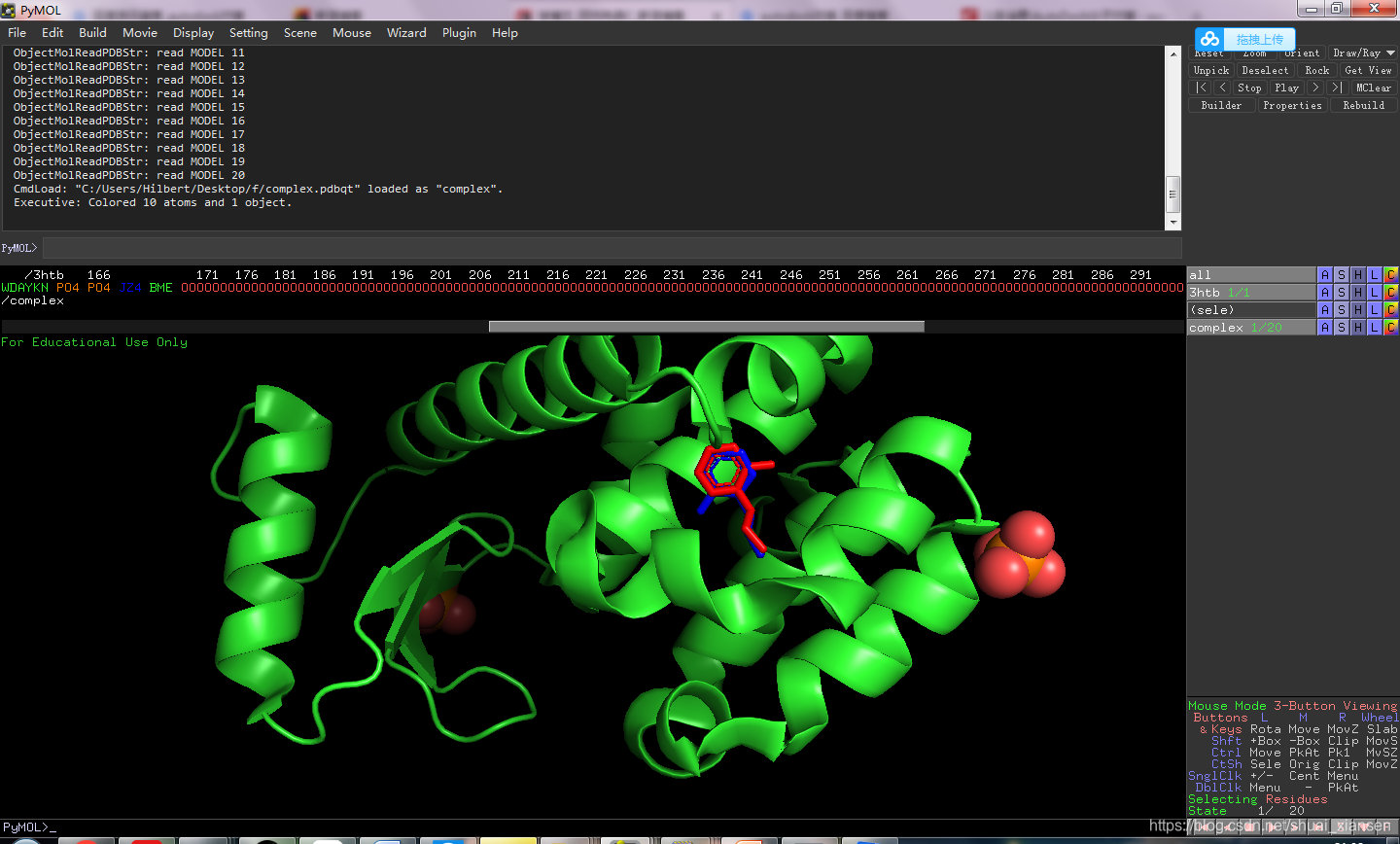
红色是对接之后的配体,蓝色是复合物中的配体,对接叠合较好,退出,对接完成

















 3710
3710

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









