






简介
本课程[1]介绍 Bioconductor 中的 ChIPseq 分析。该课程由 4 个部分组成。这将引导您完成正常 ChIPseq 分析工作流程的每个步骤。它涵盖比对、QC、peak calling、基因组富集测试、基序富集和差异 ChIP 分析。
课程材料和练习可在 https://rockefelleruniversity.github.io/Intro_To_R_1Day/ 上以呈现的 HTML 形式查看。
环境准备
IGV
IGV 可以从 BROAD 网站安装。 》 https://www.broadinstitute.org/igv/
MACS2
MACS2[2] 没有 R 包,但 MACS2 可在适用于 Linux 或 MacOS 的 Anaconda 包存储库中找到。安装 MACS2 的最简单方法是使用 R 包 Herper。 Herper 允许您从 R 中管理和安装 Anaconda 包。
BiocManager::install("Herper")
library(Herper)
安装 Herper 后,您可以使用 install_CondaTools 函数安装 MACS2。在幕后,Herper 将安装最小版本的 conda(称为 miniconda),然后创建一个新环境来安装 MACS2。当您运行该函数时,它会打印出 MACS2 的安装位置。
env 参数是您要为创建的环境指定的名称。 pathToMiniConda 指定您要安装 Miniconda 的位置,以及所有 conda 工具(如 MACS2)。
install_CondaTools(tools="macs2", env="PeakCalling_analysis", pathToMiniConda="/path/to/install")
R
见
RStudio
见
包
-
课程包
install.packages('BiocManager')
BiocManager::install('RockefellerUniversity/RU_ATACseq',subdir='atacseq')
-
来自 CRAN 和 Bioconductor
install.packages('BiocManager')
BiocManager::install('methods')
BiocManager::install('ggplot2')
BiocManager::install('rmarkdown')
BiocManager::install('ashr')
BiocManager::install('ChIPQC')
BiocManager::install('DiffBind')
BiocManager::install('ShortRead')
BiocManager::install('DESeq2')
BiocManager::install('limma')
BiocManager::install('BSgenome.Mmusculus.UCSC.mm10')
BiocManager::install('Rsubread')
BiocManager::install('Rbowtie2')
BiocManager::install('R.utils')
BiocManager::install('Rsamtools')
BiocManager::install('rtracklayer')
BiocManager::install('GenomicRanges')
BiocManager::install('TxDb.Mmusculus.UCSC.mm10.knownGene')
BiocManager::install('TFBSTools')
BiocManager::install('org.Mm.eg.db')
BiocManager::install('GenomeInfoDb')
BiocManager::install('ChIPseeker')
BiocManager::install('ggupset')
BiocManager::install('TxDb.Hsapiens.UCSC.hg38.knownGene')
BiocManager::install('GSEABase')
BiocManager::install('TxDb.Hsapiens.UCSC.hg19.knownGene')
BiocManager::install('org.Hs.eg.db')
BiocManager::install('tracktables')
BiocManager::install('goseq')
BiocManager::install('rGREAT')
BiocManager::install('GO.db')
BiocManager::install('JASPAR2020')
BiocManager::install('motifmatchr')
BiocManager::install('clusterProfiler')
BiocManager::install('enrichplot')
BiocManager::install('msigdbr')
BiocManager::install('ggnewscale')
BiocManager::install('knitr')
BiocManager::install('testthat')
BiocManager::install('yaml')
内容
Part_1
本节介绍在Bioconductor中对ChIPseq数据的分析。会话部分:
-
在 R 中预处理 ChIPseq 数据 -
数据比对 -
为可视化创建 bigWig
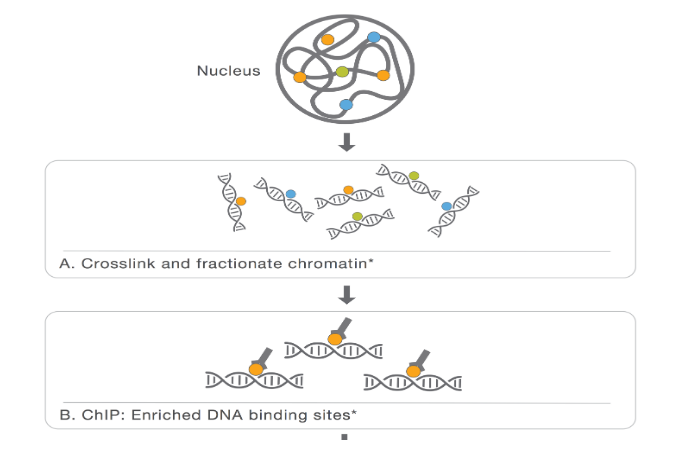
Part_2
本节涵盖更深入的 ChIPseq QC 和 MACS2 calling peaks 。会话部分:
-
R 中 ChIPseq 数据的质量控制
-
peak calling概述 -
峰的注释
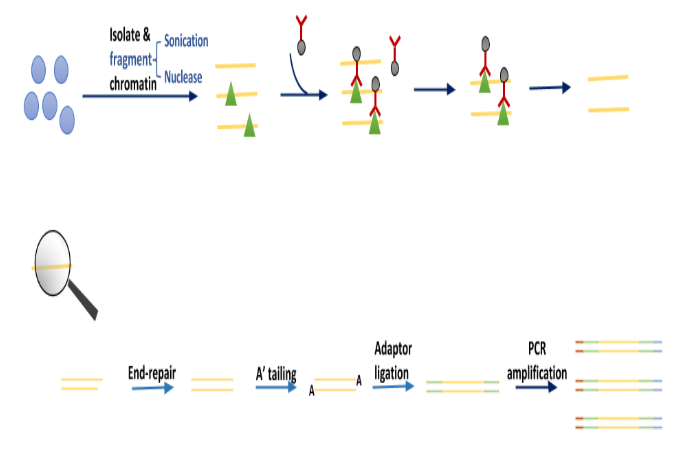
Part_3
本节介绍Bioconductor Session部分对ChIPseq数据的分析:
-
TF靶标的功能富集分析 -
与 GREAT 服务器的 R 接口 -
使用 Meme-ChIP 富集合

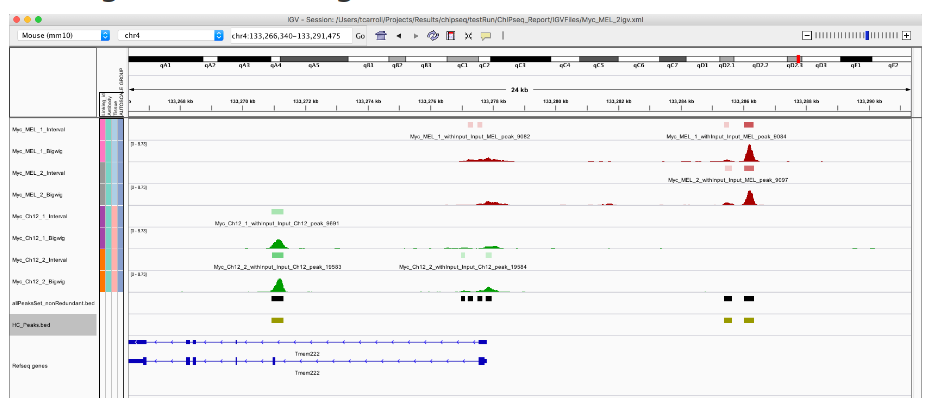
Part_4
本节介绍Bioconductor Session部分对ChIPseq数据的分析:
-
鉴定重复的、高置信度的峰 -
查找条件特有和共有的峰值 -
Differential ChIP-seq
参考资料
Source: https://rockefelleruniversity.github.io/RU_ChIPseq/
[2]MACS: https://macs3-project.github.io/MACS/
本文由 mdnice 多平台发布















 1万+
1万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









