






 本文详细介绍了ChIP-seq、DAP-seq、ATAC-seq和CUT&Tag等技术在生物学研究中的作用,重点讲解了如何利用IGV工具进行转录因子结合、组蛋白修饰等信号的可视化。指南包括如何导入基因组、注释文件,以及如何设置和调整track以获得理想的可视化效果。
本文详细介绍了ChIP-seq、DAP-seq、ATAC-seq和CUT&Tag等技术在生物学研究中的作用,重点讲解了如何利用IGV工具进行转录因子结合、组蛋白修饰等信号的可视化。指南包括如何导入基因组、注释文件,以及如何设置和调整track以获得理想的可视化效果。
背景介绍
你是不是经常在文献中看到下面这种图,好奇如何做的?在发表ChIP-seq/DAP-seq/ATAC-seq/CUT&Tag文章时,想在文章中放几张目标基因的可视化图?
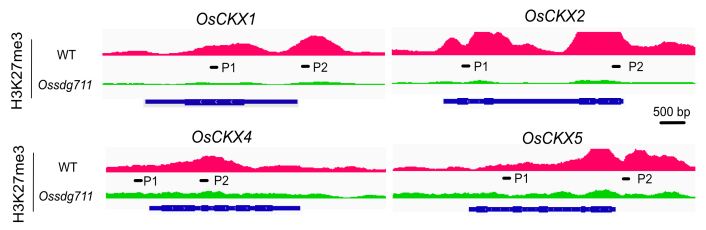
项目文章 | Plant Physiology 华南农业大学揭示组蛋白修饰调节水稻器官大小的表观遗传机制
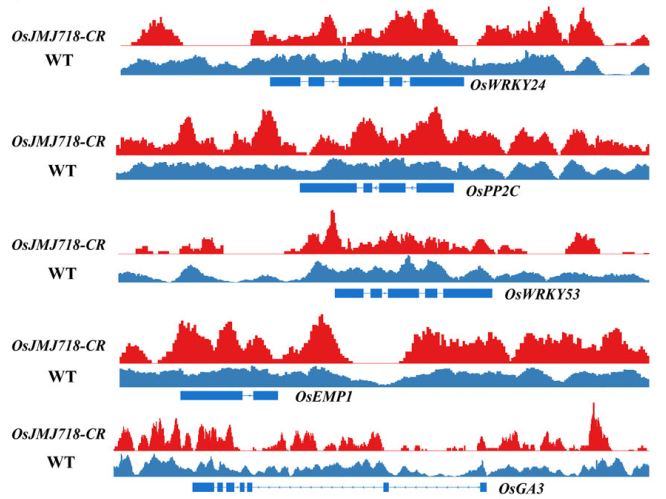
喜讯!ChIP-seq再立功,组蛋白去甲基化酶在水稻种子的表观调控机制见刊The Plant Journal
要做此图之前,我们首先得搞清楚两个问题:
1、这个图是干嘛用的?
2、怎么看?
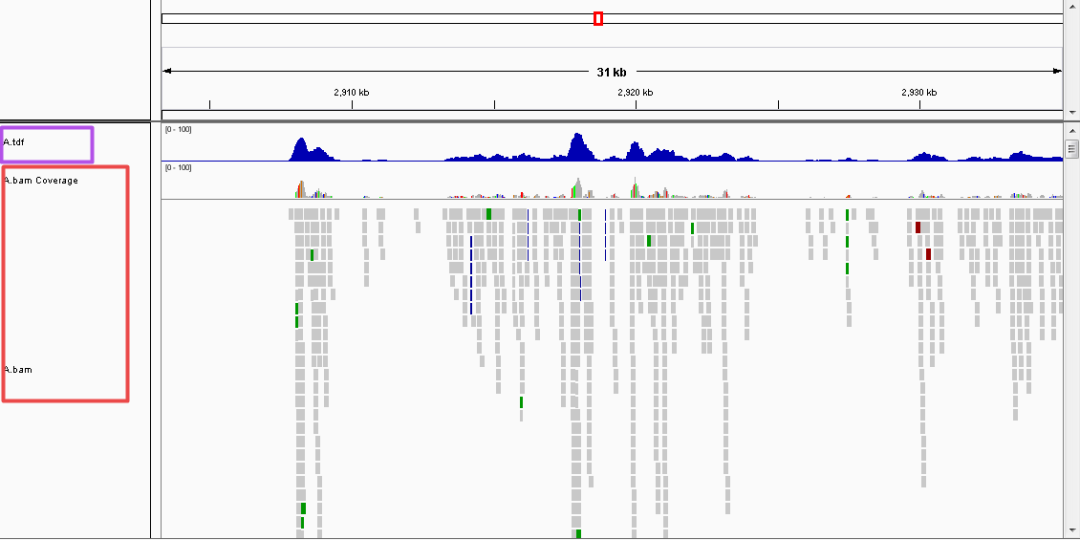
答案是:此图是测序reads的可视化,利用reads和参考基因组比对之后的结果,用来展示目标基因上的转录因子结合/组蛋白修饰/染色质开放性的。
鉴定转录因子在基因组上结合常用的技术手段是ChIP-seq/DAP-seq,检测组蛋白修饰的全基因组分布用ChIP-seq/CUT&Tag,而对染色质可及性的检测则用ATAC-seq。ChIP-seq/DAP-seq/CUT&Tag的原理都可以归纳为目标蛋白在基因组某个位点结合后,含有该位点的DNA片段将会被选择性富集;测序之后,来自于此DNA片段的reads较其他非蛋白结合的DNA片段reads多,在数据可视化上呈现一个堆集起来的山峰(在计算机概念上叫“peak”)。ATAC-seq的原理则是利用Tn5转座酶特异性识别染色质开放区并酶切该区域,这样在测序之后,来自于染色质开放区的reads也会较非染色质开放区的reads多,形成peak。
IGV基础知识
在掌握上面这些基础知识之后,就用来到今天的重点,怎么画这幅图?如果你有生信基础和环境,推荐你用R包Gviz。如果你没有生信基础,那就给你推荐今天的主角IGV。
IGV(Integrative Genomics Viewer),一款便捷的交互式使用工具,用于基因组数据的可视化,支持Windows/MacOS/Linux多平台使用,同时支持FASTA/GFF/GTF/BAM/BED/TDF/bedGraph/BEDPE/bigBED/bigGenePred/bigNarrowPeak/bigWig……多种格式文件输入。一般,我们常用到的文件格式主要是FASTA/GFF/GTF/BAM/BED/TDF/bigWig。
◆ FASTA:或称.fasta或.fa文件,指定导入参考基因组的参考序列。FASTA文件中的序列名称是出现在IGV工具栏的染色体下拉列表中的染色体名称。IGV根据染色体的名称对它们进行排序,而不是它们在FASTA文件中的顺序。
◆ GFF/GTF:通用特征格式(GFF)文件是一个简单的制表符分隔的文本文件,用于描述基因组特征(基因结构:基因起始和终止位置,外显子、内含子所在位置以及基因的转录方向)。IGV支持GFF2、GFF3和GTF文件格式,GFF3是最常用的。
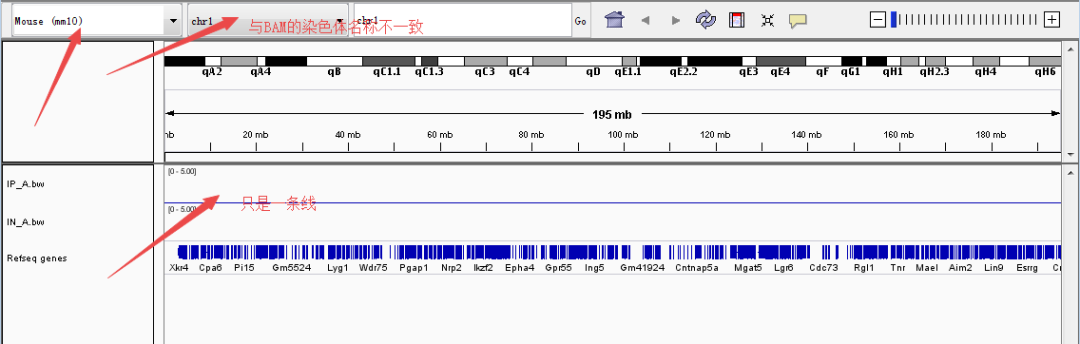
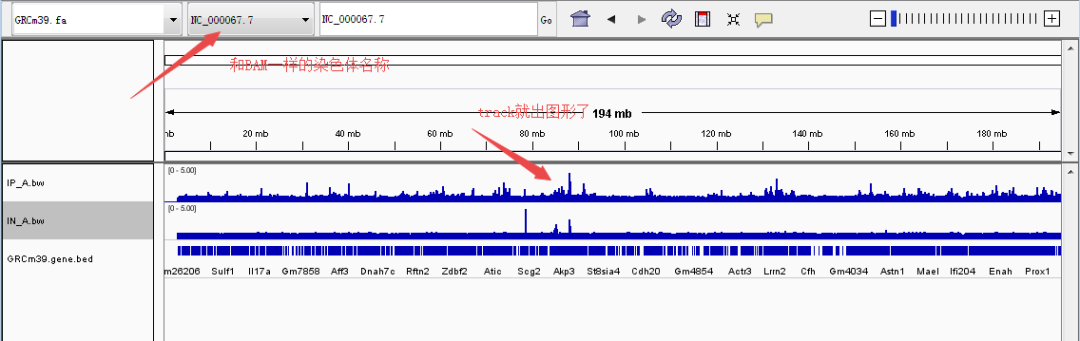
◆ BAM:BAM(.bam)文件是SAM文件的二进制版本,SAM文件(.sam)是一个包含序列比对数据的制表符分隔的文本文件,IGV也支持BAM格式。值得注意的有两点,一是BAM和SAM都得按位置排序并索引,排序和索引可以用igvtools完成;二是参考基因组和SAM/BAM的染色体名称必须一致(很多人很容易忽视这点,导致文件导入后没有图像,只是一条线)。
◆ BED:BED(.bed)是一个用于定义特征(可以是基因、SNP位点、甲基化位点、peak)轨迹以制表符分隔的文本文件,它可以有任意文件扩展名,但建议是.bed。
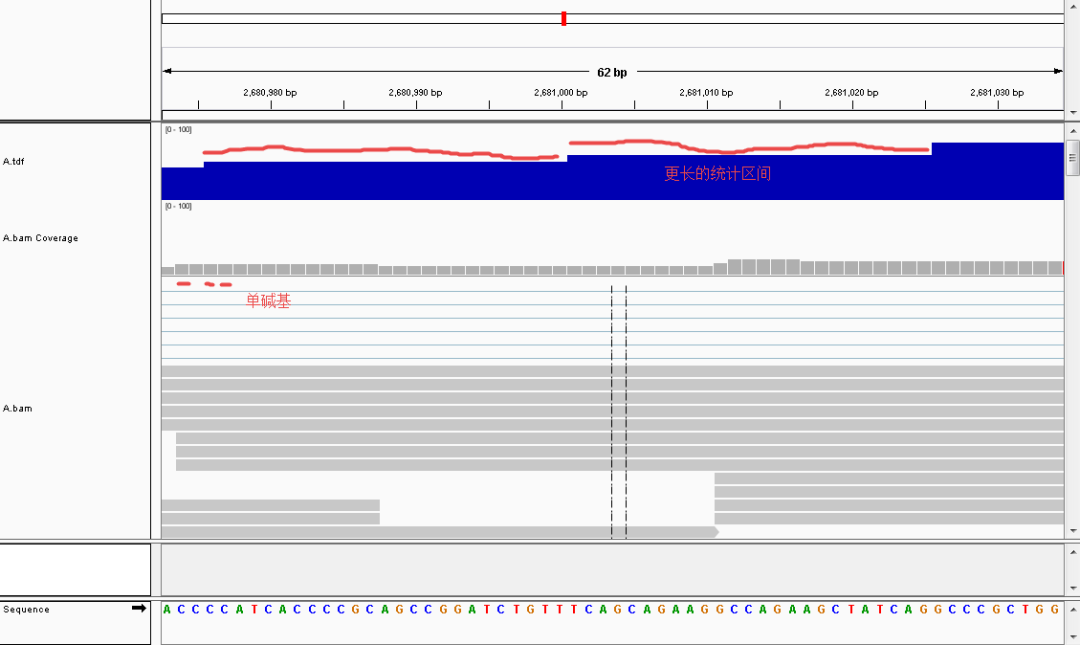
◆ TDF:TDF(.tdf)是一个二进制文件,用窗口的方式来记录测序深度信息。相比BAM文件,TDF文件会小很多,导入和查看也更快速。可以通过igvtools来生成TDF文件。下图清晰表明了两者的区别,在较小的分辨率下,bamcoverage基本和tdf无异;但当分辨率较大的时候,可以看出bam是以单碱基存储深度的,而tdf是以区域内储存平均深度的。
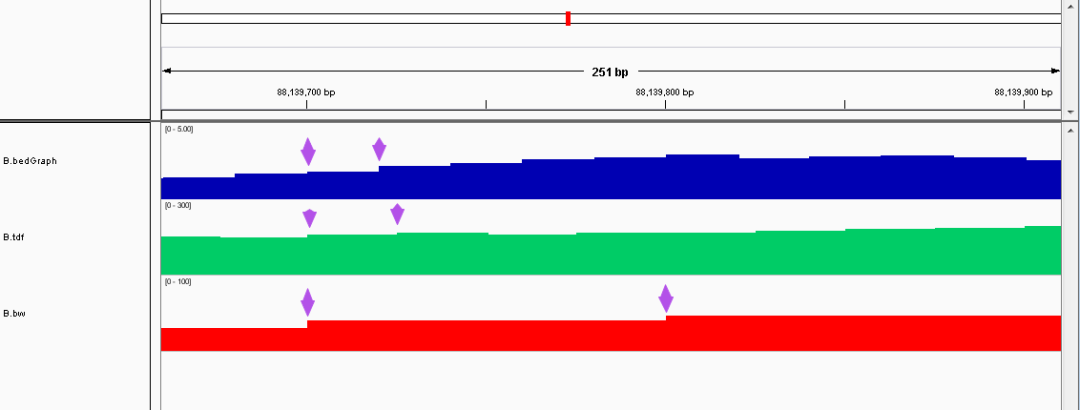
◆ bigWig:和TDF类似,只是统计区间相较于TDF更大。类似的文件还有bedGraph,统计区域比TDF还小。下图展示了三种文件的区别,在小分辨率下几乎没差异,在大分辨率下就会看出细节差异。因此,在文件大小上,bedGraph > TDF > bigwig。一般以bigWig(.bw)上传到数据库。



IGV的使用
第一步:IGV安装
上IGV官网,下载对应电脑系统的程序,按照提示安装即可。下载链接:https://igv.org/doc/desktop/#DownloadPage/
第二步:IGV使用
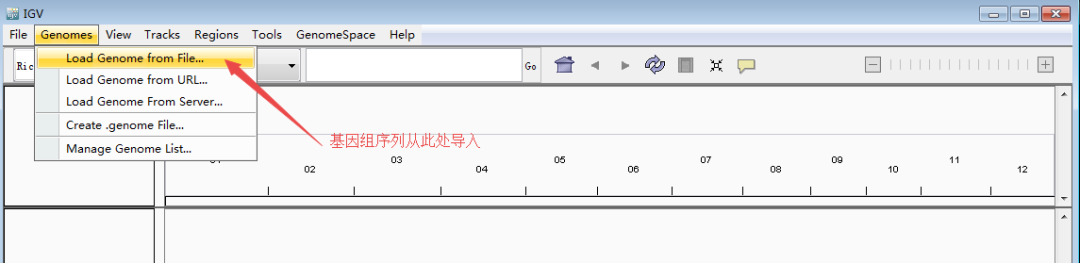
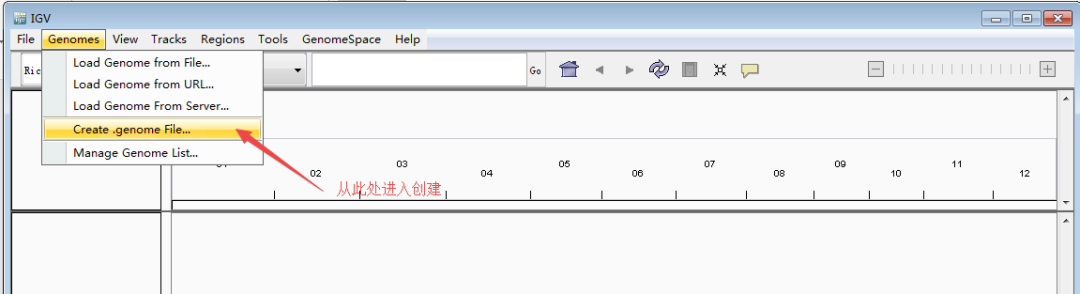
首先,导入基因组。从IGV导航栏里的Genomes >> Load Genome from File进入;
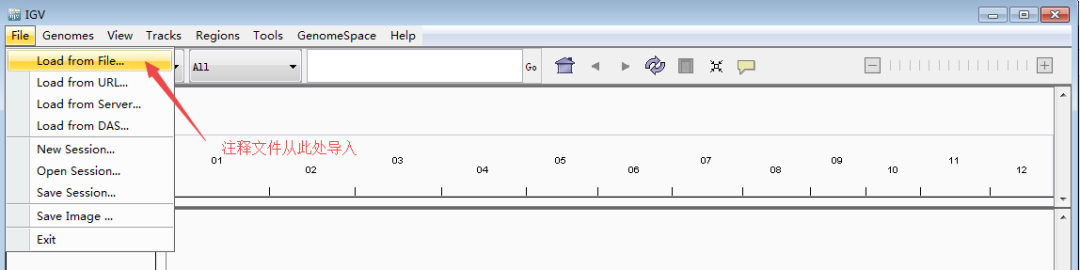
接着,导入注释文件。从导航栏里的File >> Load from File进入;
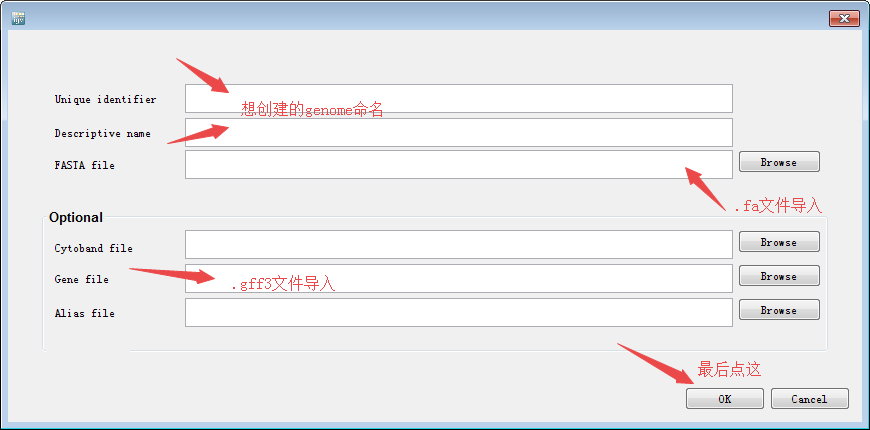
当然,你也可以选择创建一个genome文件,将序列文件和注释文件合一,下次就不用再分别导入两个文件了。创建方法为:从导航栏里的Genomes >> Create. Genome File进入,然后在对话框里依次输入待创建基因组的命名,导入序列文件和注释文件,最后点击确认,设置好存储位置即可。
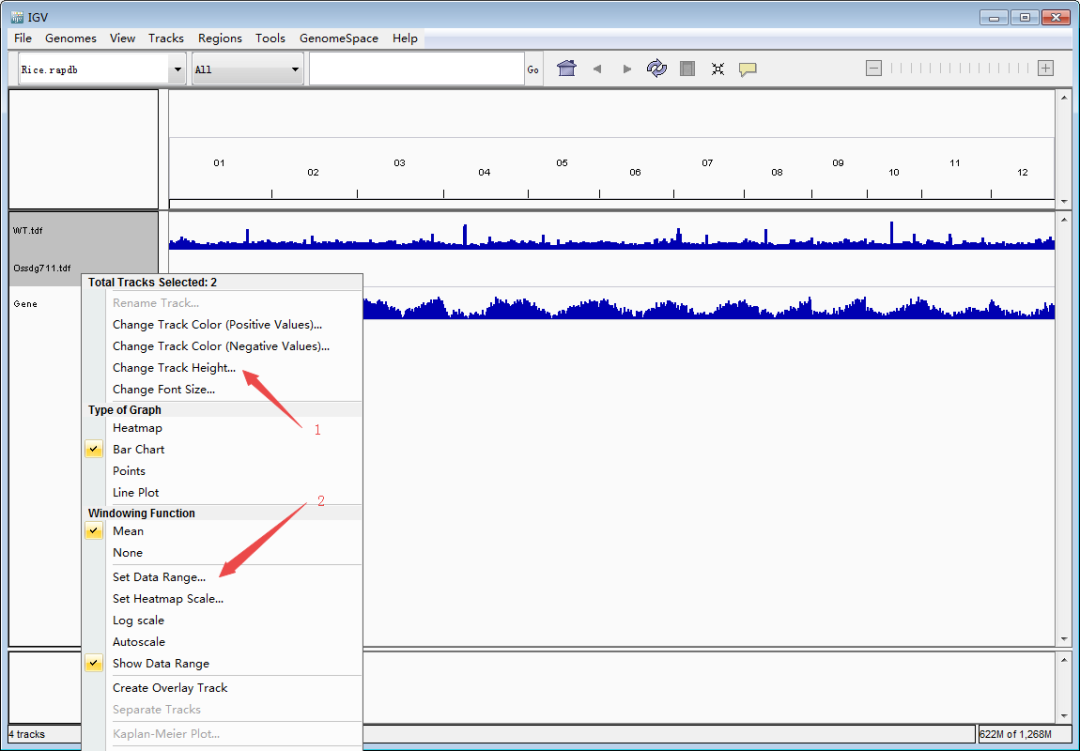
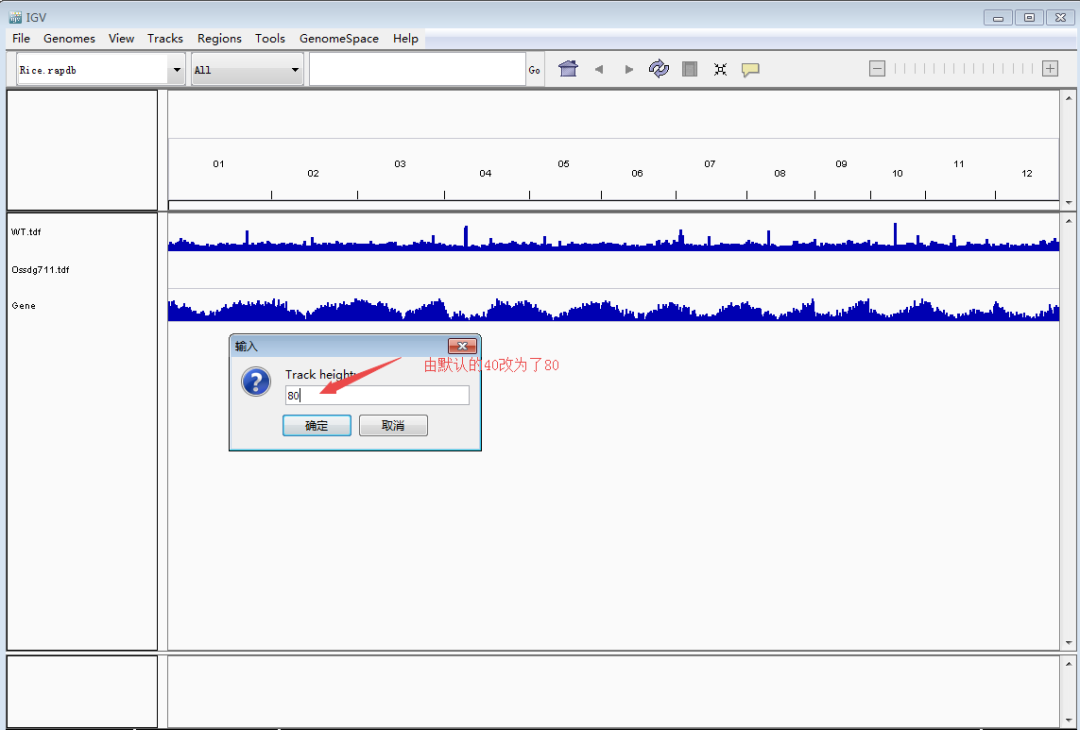
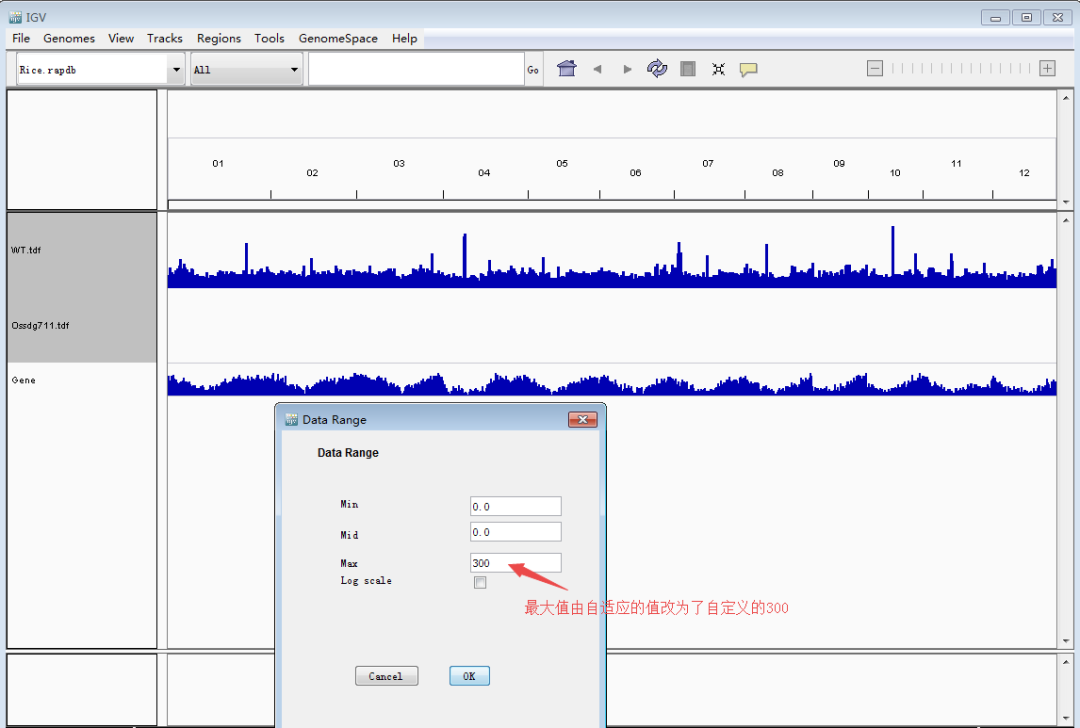
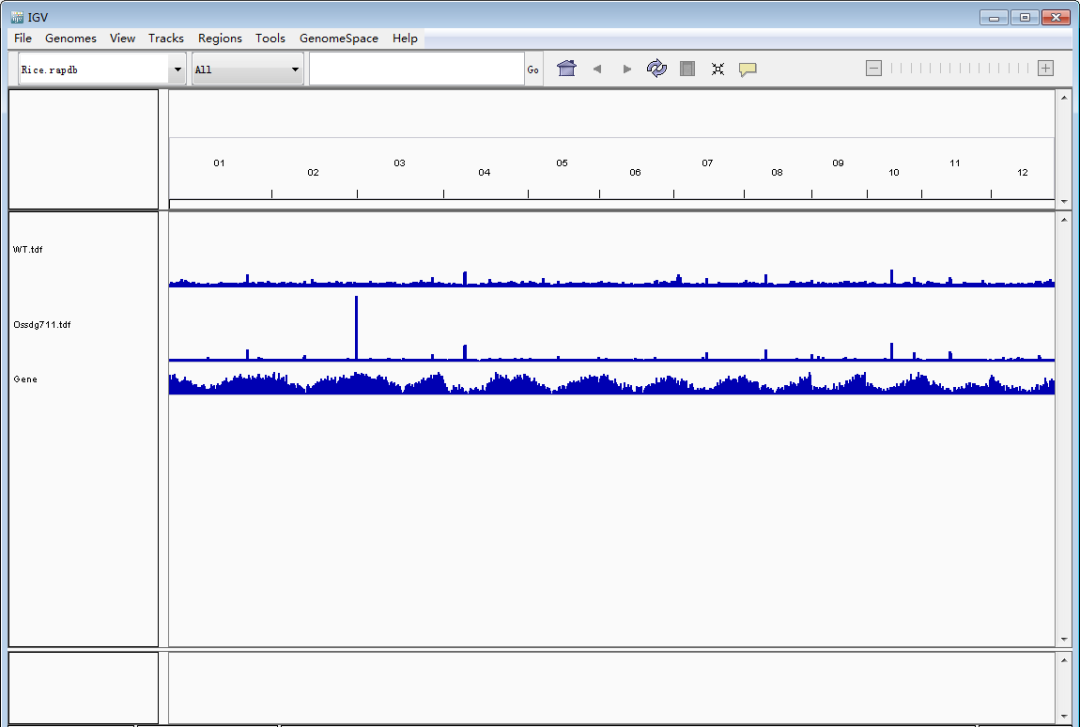
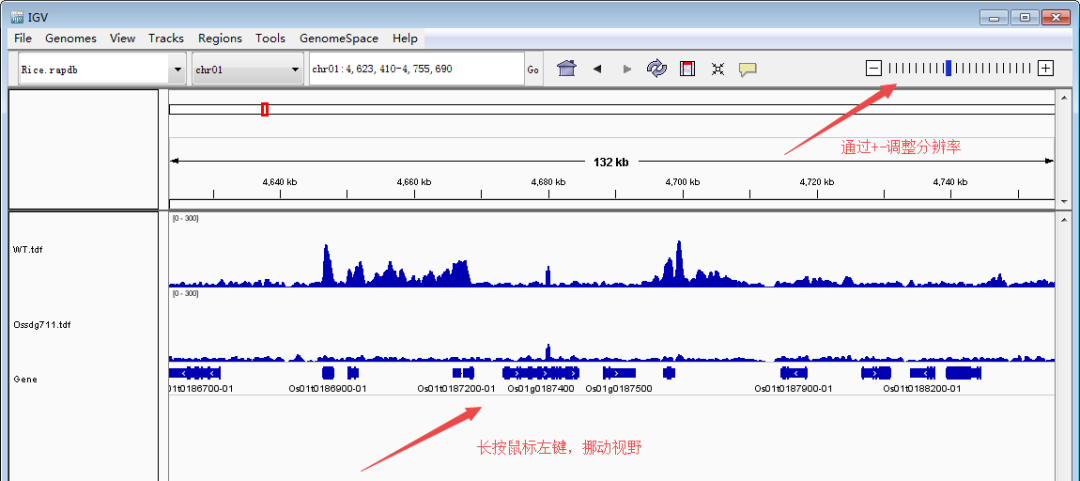
接下来是重点,导入待看的track文件(TDF/bigwig/bedgraph)。单击鼠标左键选择track(ctrl+单机左键可同时选择多个track),然后单击鼠标右键调出设置栏,调整track高度和data的范围,使峰型图在视野里看起来更合适。Tips:每个基因上的强度存在差异,因此data的range要根据目标基因设定,可以多次设定。图形没完全显示,就把值调大点;图形太矮,就把值调小一点。总之一点,怎么好看怎么来,调到自己满意即可。
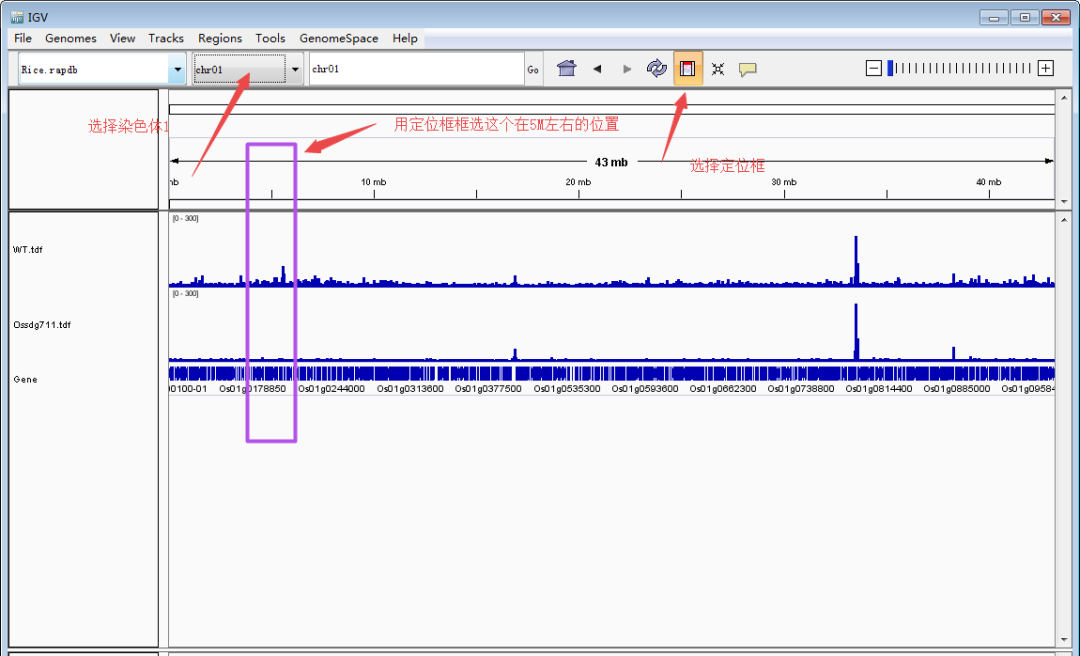
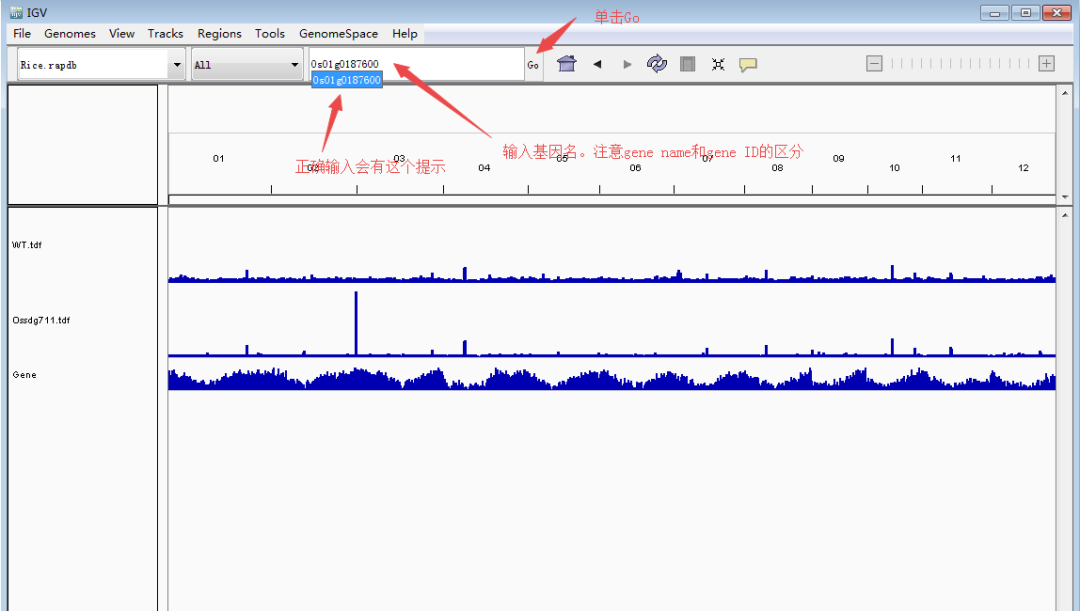
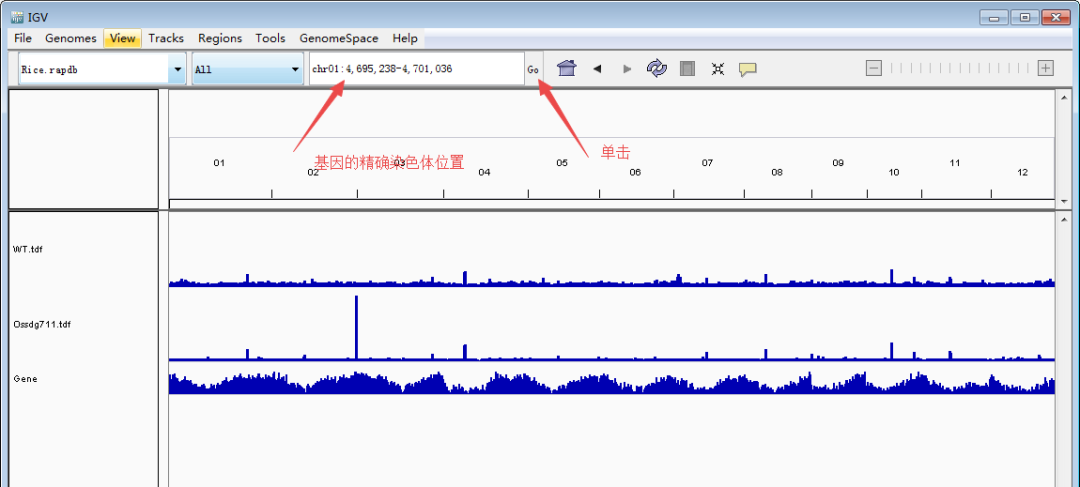
随后,定位到目标基因。下面以本文第一幅图的OsCKX1(Os01g0187600:chr01:4,695,238-4,701,036)为例展示。主要有两种方法:1)大概定位法:根据基因的大概位置,下拉染色体框,选择对应的染色体,然后缩小区间。比如OsCKX1在chr1上的大概4.7M的位置,先用定位框选择5Mb左右的一个大框,然后再在4.7Mb再次精细框选。2)直接输入法:在输入框里面输入基因(注意基因名和基因ID的区分)或者基因的精确染色体定位,点击Go就可以直接跳转到目标基因了。
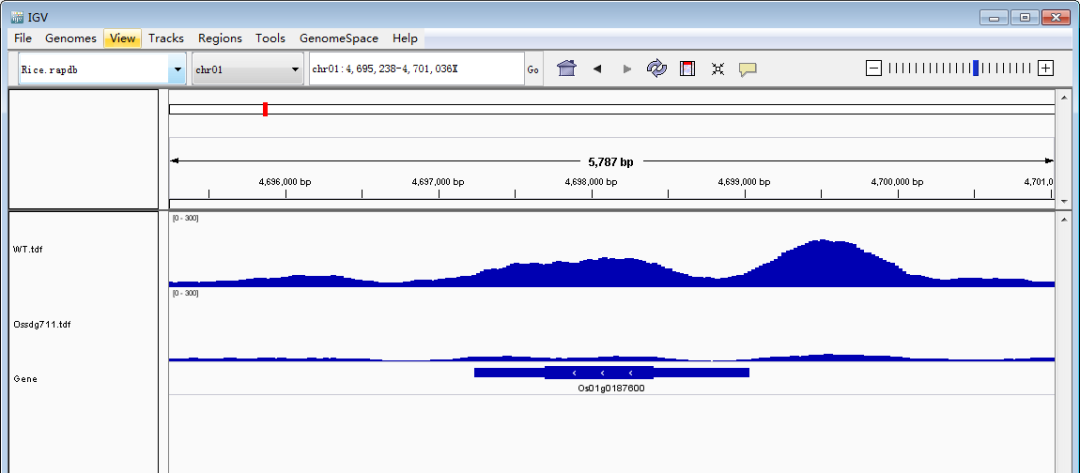
以上两种方法都可以定位到目标基因,随后就是图像美化,我们按照发表文章里的色泽。将WT的track改成粉色,Ossdg711的track改成绿色。和调整track的高度和数据值一样,先调出设置栏,点击Change track Color,选择想要的颜色。Tips:不同track设置不同颜色时,要一个一个track调整。
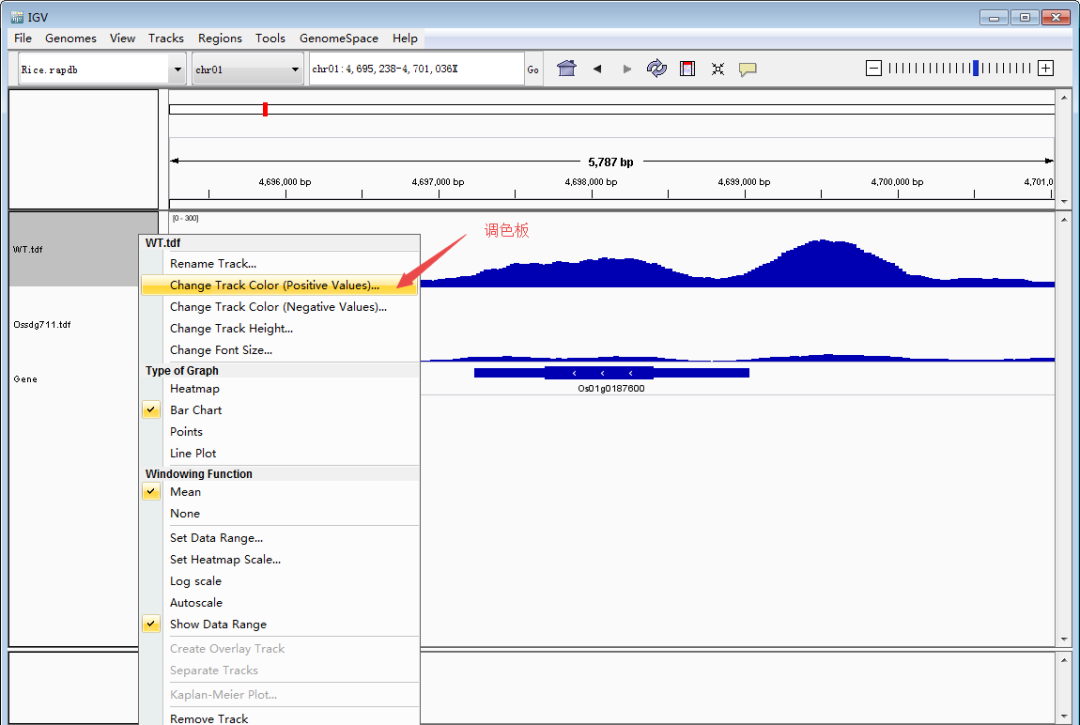
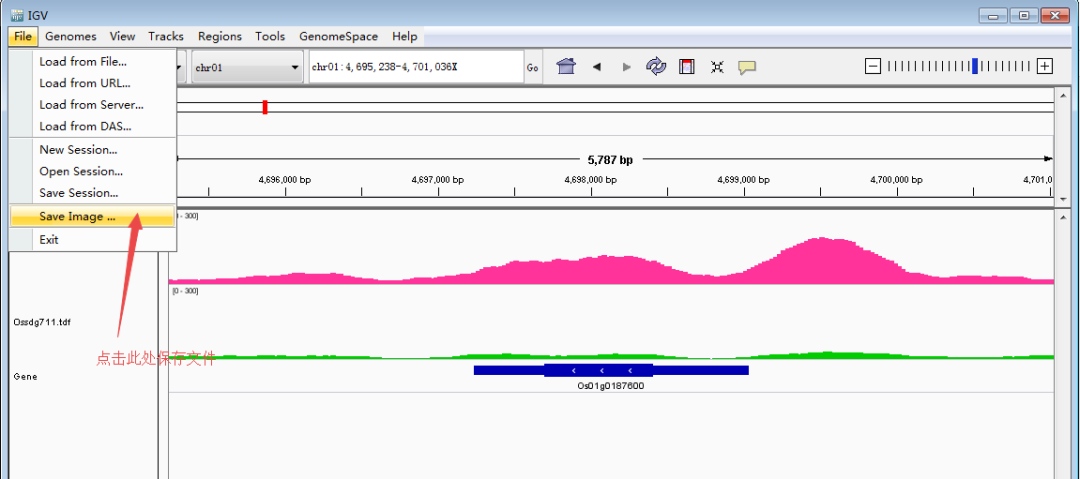
调整好了图像之后,从File >> Save Image保存图像,以矢量图.svg格式存储,方便后面用AI编辑。
后 记
如果您觉得自己一个一个基因编辑美化太麻烦,爱基百客生物可以提供ChIP-seq/DAP-seq/ATAC-seq/CUT&Tag等全套技术服务,提供可视化文件供您检索基因。此外,我们的生信团队还能提供您挑选后的指定基因的可视化绘图服务。欢迎与我们联系。

















 2820
2820

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









