







再不更新就要忘记学习了,继续坚持分享,今天分享的是单细胞批次效应校正方法的fastMNN算法。
MNN原理介绍
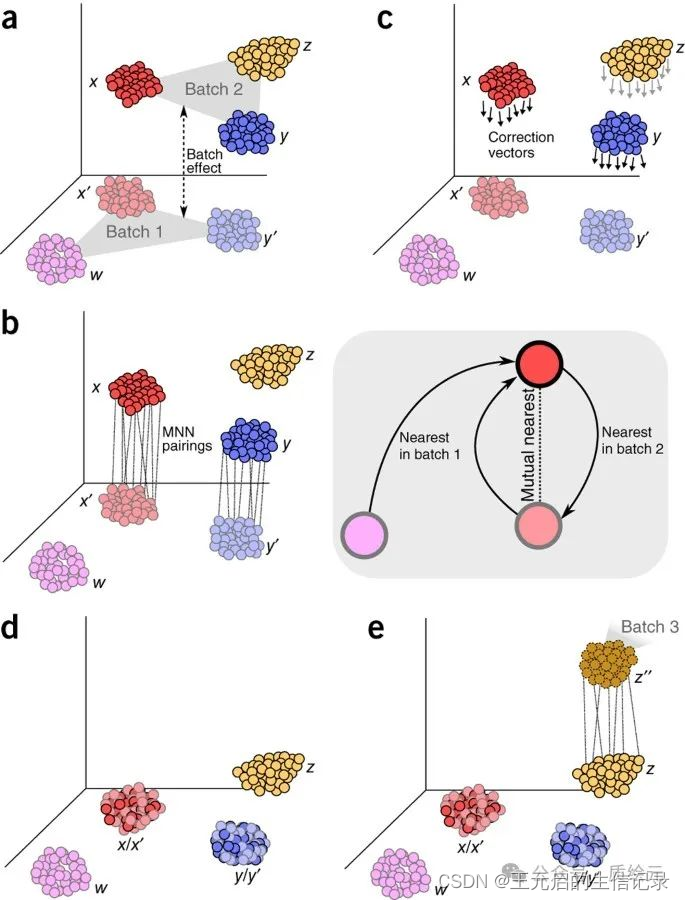
先从MNN讲起,MNN是Haghverdi等人提出的一种批次校正算法,由R语言的batchelor包实现。算法原理及性能评测可自行阅读论文:Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors。大部分批次效应校正方法错误地假设细胞群的组成在各个批次之间是已知的或相同的。而MNN方法不依赖于各个批次之间预定义或相等的群体组成;相反,它只要求各个批次之间共享一个群体子集。
MNN使用假设:(i)至少有一个细胞群同时存在于两个批次中,(ii)批次效应几乎与生物子空间正交,(iii)批次效应变化远小于不同细胞类型之间的生物效应变化。
MNN pair:
首先根据两个批次样本的表达矩阵计算细胞之间的余弦距离(常用来表示单细胞转录特征相似性的度量值,可以在一定程度上屏蔽测序深度和捕获效率不同造成的差异)
然后为Batch1样本中的细胞(记作Cell-i)在Batch2样本寻找k个余弦距离最近的细胞集(记作Cell-set-i)
同样为Batch2样本中的细胞(记作Cell-j)在Batch1样本寻找k个距离最近的细胞集(记作Cell-set-j)
如果Cell-i在Cell-Set-j中,恰好Cell-j也在Cell-Set-i中,那么Cell-i与Cell-j就是相互最近邻细胞对(即MNN pair)
MNN pair 中细胞之间表达值的差异提供了批次效应的估计值,通过对许多这样的MNN pair对取平均值可以使其更加精确。从估计的批次效应中获得校正向量,并将其应用于表达值以执行批次校正
fastMNN原理介绍
fastMNN是MNN的升级版,主要改动是fastMNN采用PCA降维之后的低维空间计算细胞之间的距离,而MNN直接使用原始表达矩阵计算细胞之间的距离,因此分析速度会更快。
fastMNN简述:
对(余弦)标准化表达值执行多样本 PCA 以降低维数。
在参考批次和目标批次之间的低维空间中识别 MNN 对。
消除参考批次和目标批次中沿平均批次向量的变化。
使用局部加权校正向量,将目标批次中的细胞校正至参考值。
将更正后的目标批次与参考批次合并,然后重复下一个目标批次。
fastMNN实现:
实现方法源自:https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1850-9#Sec13。
数据:dataset4,胰腺细胞数据
代码复现:
1.使用batchelor包进行mnn,根据文献中的fastMNN进行分析
library(Seurat)
library(batchelor)
expr_mat_mnn <- readRDS(file = "C:/Users/Wang/Documents/单细胞学习/单细胞蛋白组学/代码/MNNanalysis/data/pancreas_v3_files/pancreas_expression_matrix.rds")
metadata <- readRDS(file = "C:/Users/Wang/Documents/单细胞学习/单细胞蛋白组学/代码/MNNanalysis/data/pancreas_v3_files/pancreas_metadata.rds")
######数据前处理
b_seurat <- CreateSeuratObject(expr_mat_mnn, meta.data = metadata, project = "MNNcorrect_benchmark")
normData = T
norm_method = "LogNormalize"
scale_factor = 10000
batch_label = "tech"
celltype_label = "celltype"
#####对数据进行归一化
if (normData) {
b_seurat <- NormalizeData(object = b_seurat, normalization.method = norm_method, scale.factor = scale_factor)
} else {
b_seurat@data = b_seurat@raw.data
}
b_seurat <- ScaleData(object = b_seurat)
b_seurat <- FindVariableFeatures(object = b_seurat)
b_seurat <- ScaleData(object = b_seurat)
b_seurat <- RunPCA(object = b_seurat)
b_seurat <- FindNeighbors(object = b_seurat, dims = 1:30)
b_seurat <- FindClusters(object = b_seurat)
b_seurat <- RunUMAP(object = b_seurat, dims = 1:30)
DimPlot(object = b_seurat, reduction = "umap")
meta_data <- b_seurat@meta.data
data_filtered<- b_seurat@assays$RNA$scale.data
#split data into batches
data_list <- list()
meta_list <- list()
list_batch <- unique(meta_data[,batch_label])
for (i in 1:length(list_batch)) {
selected <- row.names(meta_data)[which (meta_data[, batch_label] == list_batch[i])]
data_list[[i]] <- data_filtered[,selected]
meta_list[[i]] <- meta_data[selected, ]
}
b1_data <- data_list[[1]]
b2_data <- data_list[[2]]
b1_meta <- meta_list[[1]]
b2_meta <- meta_list[[2]]
batch_data<- list(b1_data, b2_data, b1_meta, b2_meta)
#####函数实现####
call_mnn(data_list,meta_list, batch_label, celltype_label)
#####call_mnn函数
call_mnn <- function(batch_data, batch_label, celltype_label, npcs = 20,
plotout_dir = "", saveout_dir = "",
outfilename_prefix = "",
visualize = T, save_obj = T)
{
#plotting
k_seed = 10
tsne_perplex = 30
tsneplot_filename = "_fastmnn_tsne"
mnn_out_filename = "_fastmnn_out"
metadata_out_filename = "_fastmnn_metadata_out"
pca_filename = "_fastmnn_pca"
b1_data <- batch_data[[1]]
b2_data <- batch_data[[2]]
b1_meta <- batch_data[[3]]
b2_meta <- batch_data[[4]]
metadata <- rbind(b1_meta, b2_meta)
##############################################run
t1 = Sys.time()
cosd1 <- cosineNorm(as.matrix(b1_data))
cosd2 <- cosineNorm(as.matrix(b2_data))
pcs_total <- multiBatchPCA(cosd1, cosd2) #defaut npca = 50
out_mnn_total <- fastMNN(pcs_total[[1]], pcs_total[[2]], pc.input = TRUE)
t2 = Sys.time()
print(t2-t1)
######################################save
saveRDS(out_mnn_total, file=paste0(saveout_dir, outfilename_prefix, mnn_out_filename, ".RDS"))
corrected_pca_mnn <- as.data.frame(out_mnn_total$corrected)
rownames(corrected_pca_mnn) <- colnames(cbind(b1_data,b2_data))
cells_use <- rownames(corrected_pca_mnn)
metadata <- metadata[cells_use,]
corrected_pca_mnn[rownames(metadata), batch_label] <- metadata[ , batch_label]
#corrected_pca_mnn[rownames(metadata), 'batch'] <- metadata[ , "batch"]
corrected_pca_mnn[rownames(metadata), celltype_label] <- metadata[ , celltype_label]
write.table(corrected_pca_mnn, file=paste0(saveout_dir, outfilename_prefix, pca_filename, ".txt"), quote=F, sep='\t', row.names = T, col.names = NA)
saveRDS(metadata, file = paste0(saveout_dir, outfilename_prefix, metadata_out_filename, ".RDS"))
#write.table(metadata, file=paste0(saveout_dir, outfilename_prefix, metadata_out_filename, ".txt"), quote=F, sep="\t", row.name=TRUE, col.name=NA)
if (visualize) {
set.seed(10)
out_tsne <- Rtsne(out_mnn_total$corrected, check_duplicates=FALSE, perplexity = 30)
tsne_df<- as.data.frame(out_tsne$Y)
rownames(tsne_df) <- colnames(cbind(b1_data,b2_data))
dim(tsne_df)
colnames(tsne_df) <- c('tSNE_1', 'tSNE_2')
tsne_df$batchlb <- metadata[ , batch_label]
#tsne_df$batch <- metadata[ , "batch"]
tsne_df$CellType <- metadata[ , celltype_label]
p01 <- ggplot(tsne_df, aes(x = tSNE_1, y = tSNE_2, colour = batchlb)) + geom_point(alpha = 0.6) + theme_bw()
p01 <- p01 + labs(x='tSNE_1',y='tSNE_2',title='fast MNN')
p01 <- p01 + theme(legend.title = element_text(size=17),
legend.key.size = unit(1.1, "cm"),
legend.key.width = unit(0.5,"cm"),
legend.text = element_text(size=14),
plot.title = element_text(color="black", size=20, hjust = 0.5))
p02 <- ggplot(tsne_df, aes(x = tSNE_1, y = tSNE_2, colour = CellType)) + geom_point(alpha = 0.6) + theme_bw()
p02 <- p02 + labs(x='tSNE_1',y='tSNE_2',title='fast MNN')
p02 <- p02 + theme(legend.title = element_text(size=17),
legend.key.size = unit(1.1, "cm"),
legend.key.width = unit(0.5,"cm"),
legend.text = element_text(size=14),
plot.title = element_text(color="black", size=20, hjust = 0.5))
png(paste0(plotout_dir,outfilename_prefix,tsneplot_filename,".png"),width = 2*1000, height = 800, res = 2*72)
print(plot_grid(p01, p02))
dev.off()
pdf(paste0(plotout_dir,outfilename_prefix,tsneplot_filename,".pdf"),width=15,height=7,paper='special')
print(plot_grid(p01, p02))
dev.off()
}
}
- 使用seurat-wrappers包实现fastMNN
rm(list=ls())
remotes::install_github('satijalab/seurat-wrappers')
library(Seurat)
library(tidyverse)
library(Seurat)
library(tidyverse)
library(patchwork)
library(SeuratWrappers)
dir <- dir("GSE139324/") #GSE139324是存放数据的目录,自行下载进行分析
dir <- paste0("GSE139324/",dir)
sample_name <- c('HNC01PBMC', 'HNC01TIL', 'HNC10PBMC', 'HNC10TIL', 'HNC20PBMC',
'HNC20TIL', 'PBMC1', 'PBMC2', 'Tonsil1', 'Tonsil2')
scRNAlist <- list()
for(i in 1:length(dir)){
counts <- Read10X(data.dir = dir[i])
scRNAlist[[i]] <- CreateSeuratObject(counts, project=sample_name[i], min.cells=3, min.features = 200)
scRNAlist[[i]][["percent.mt"]] <- PercentageFeatureSet(scRNAlist[[i]], pattern = "^MT-")
scRNAlist[[i]] <- subset(scRNAlist[[i]], subset = percent.mt < 10)
}
saveRDS(scRNAlist, "scRNAlist.rds")
scRNAlist <- readRDS("scRNAlist.rds")
##为了更好展现整合效果,只使用两个样本整合
scRNAlist <- list(scRNAlist[[2]], scRNAlist[[10]])
scRNAlist <- lapply(scRNAlist, FUN = function(x) NormalizeData(x))
scRNAlist <- lapply(scRNAlist, FUN = function(x) FindVariableFeatures(x))
scRNA <- RunFastMNN(object.list = scRNAlist)
scRNA <- RunUMAP(scRNA, reduction = "mnn", dims = 1:30)
scRNA <- FindNeighbors(scRNA, reduction = "mnn", dims = 1:30)
scRNA <- FindClusters(scRNA)
p1 <- DimPlot(scRNA, group.by = "orig.ident", pt.size=0.1)
ggtitle("Integrated by fastMNN")
p2 <- DimPlot(scRNA.orig, group.by="orig.ident", pt.size=0.1)
ggtitle("No integrated")
p = p1 p2 plot_layout(guides='collect')
ggsave('fastMNN.png', p, width=8, height=4)
欢迎关注转发与交流,如有疑问请联系管理员:kriswcyYQ进行相关问题解决。















 620
620

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









