









这是GATK Best Practice系列学习文章中的一篇,本文尝试使用:
Gatk RNA -Seq spns-indels Pipeline 来分析鼻咽癌(NPT)
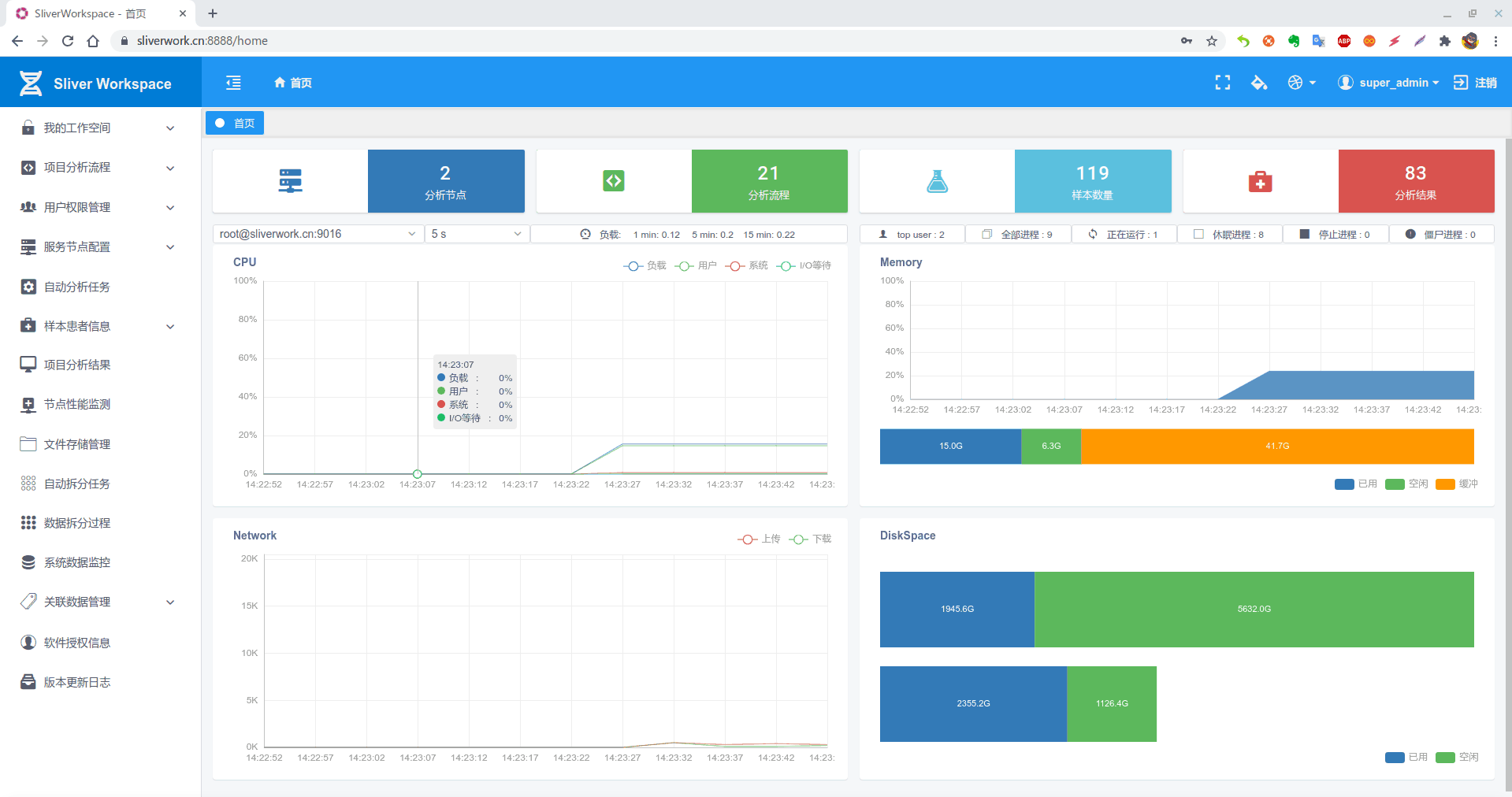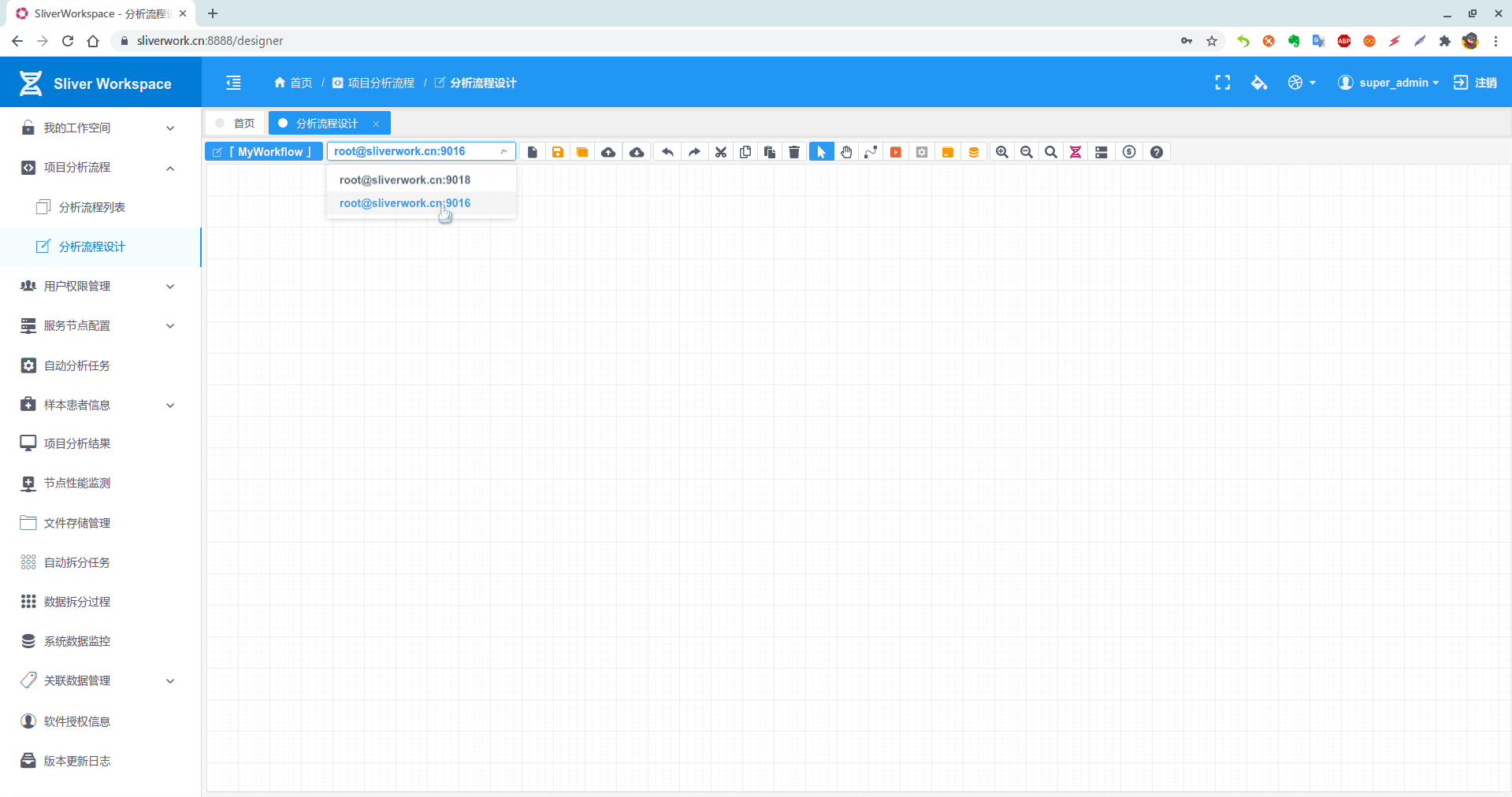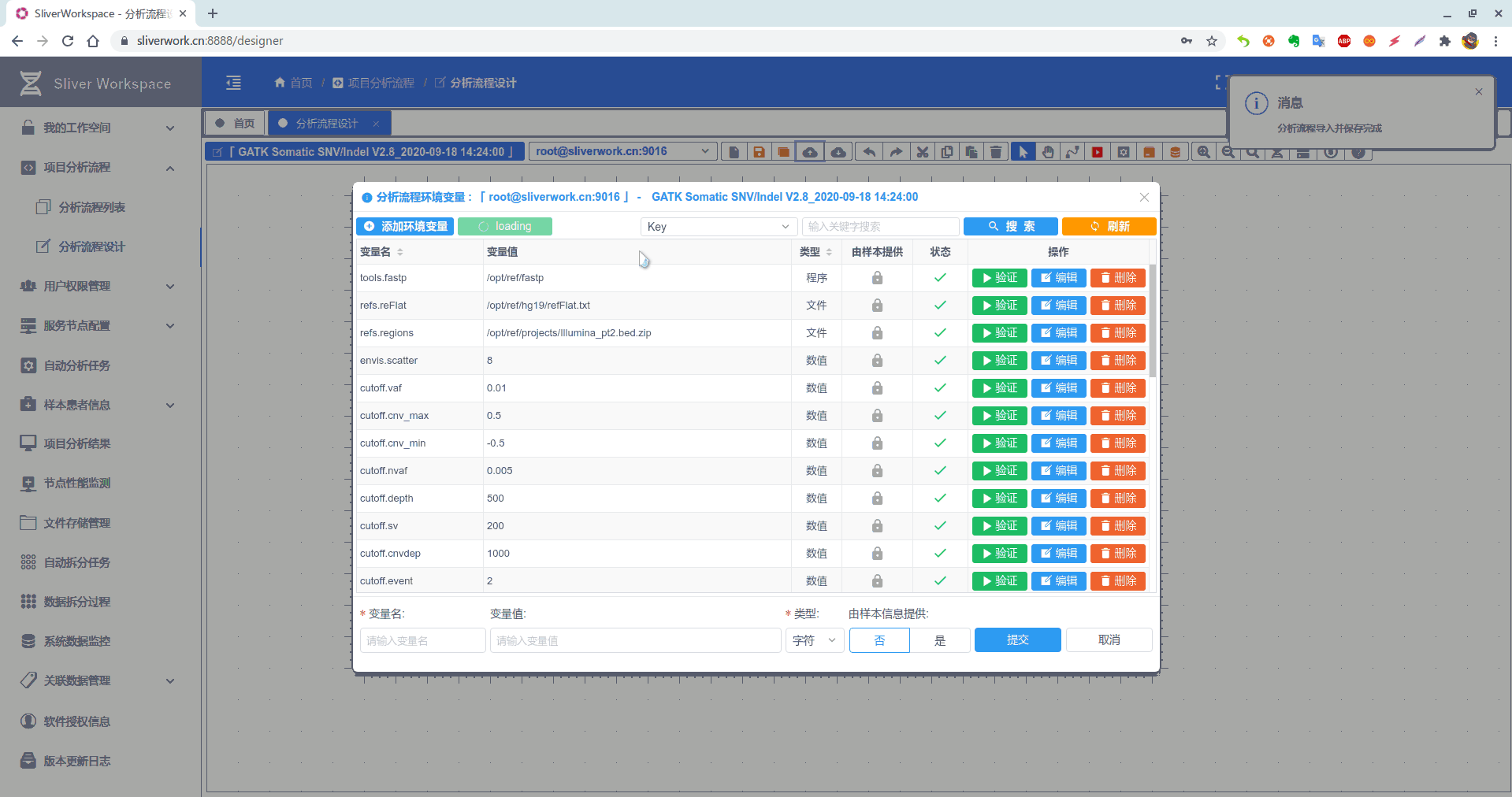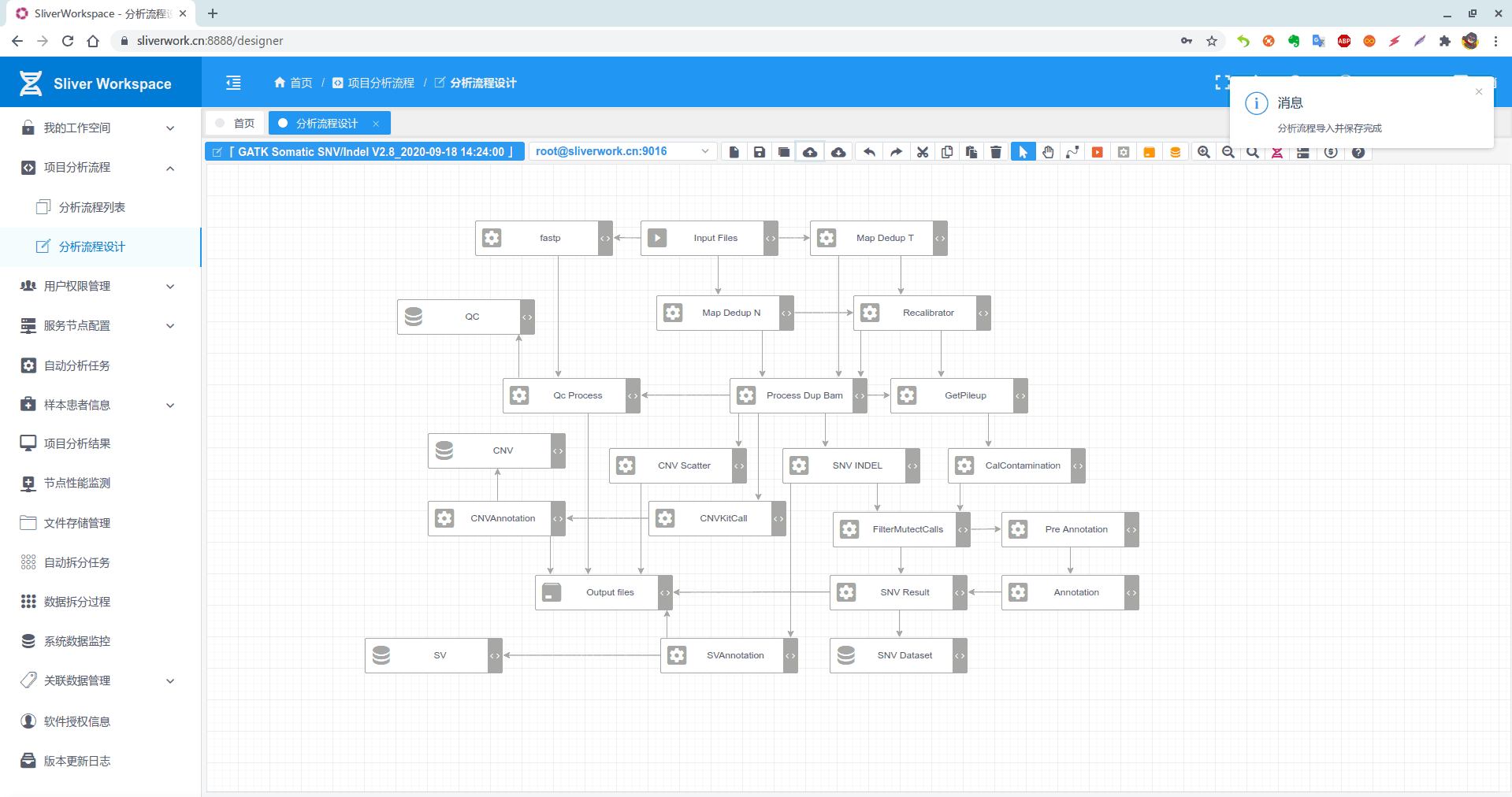
分析流程如下:

GATK版本的是这样的

数据
从NCBI上下载转录组数据,访问链接为:
https://trace.ncbi.nlm.nih.gov/Traces/study/?acc=SRP058243&o=acc_s%3Aa

第一个样本的数据下载链接如下:
| Location | Name Link |
|---|---|
| NCBI | https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR2016932/SRR2016932.1 |
| NCBI | https://sra-downloadb.st-va.ncbi.nlm.nih.gov/sos2/sra-pub-run-6/SRR2016932/SRR2016932.1 |
#下载完成后获得文件为SRR2016932.1,需要转换为fastq.gz格式,这里用到NCBI sratookit工具,请自行安装
fastq-dump -gzip --split-3 SRR2016932.1 -O ./
#得到两个fastq.gz文件
SRR2016932_1.fastq.gz
SRR2016932_2.fastq.gz
#我们将文件重命名为以符合习惯
mv SRR2016932_1.fastq.gz SRR2016932_R1.fastq.gz
mv SRR2016932_2.fastq.gz SRR2016932_R2.fastq.gz
用到的分析系统及分析流程文件
| 名称 (点击下载) | 备注 |
|---|---|
| SliverWorkspace 2.0.295368 | 提供运行控制平台/社区版 |
| GATK Germline SNP/Indel V1.2 | 分析流程文件,可以一键导入分析平台(点击查看操作) 不想复制shell的,可以使用平台一键导入流程,当然reference文件和软件还需要自己下载和安装 |
| ucsc.hg19.gtf.tar.xz | ucsc.h19.gtf ucsc.hg19.gtf.bed 从ucsc.hg19.gtf中列数据中生成的bed文件 ucsc.hg19.gtf.interval_list 使用gatk IntervalToBed工具从ucsc.hg19.gtf.bed转换来的interval文件 |
| 分析结果 | 分析结果 |
流程用到的变量(程序、reference文件和数据库、数值)
| 变量名 | 变量值 | 类型 |
|---|---|---|
| sn | SRR2016932 | 字符 |
| tools.fastqc | /opt/FastQC/fastqc | 程序 |
| tools.STAR | /opt/ref/STAR-2.7.3a/bin/Linux_x86_64_static/STAR | 程序 |
| tools.sambamba | /opt/ref/sambamba-0.7.0-linux-static | 程序 |
| tools.gatk | /opt/ref/gatk-4.1.4.1/gatk | 程序 |
| envis.read_length | 100 (测序读长) | 数值 |
| envis.threads | 32 (并发线程数) | 数值 |
| envis.scatter | 10 (HaplotypeCaller并行数) | 数值 |
| refs.hum | /opt/ref/hg19/ucsc.hg19.fa | 文件 |
| refs.gtf | /opt/ref/RNA/ucsc.hg19.gtf | 文件 |
| refs.bed | /opt/ref/RNA/ucsc.hg19.gtf.bed | 文件 |
| refs.interval | /opt/ref/RNA/ucsc.hg19.gtf.interval_list | 文件 |
| refs.1000G | /opt/ref/hg19/1000G_phase1.indels.hg19.vcf | 文件 |
| refs.mills | /opt/ref/hg19/Mills_and_1000G_gold_standard.indels.hg19.vcf | 文件 |
| refs.dbsnp | /opt/ref/hg19/dbsnp_138.hg19.vcf | 文件 |
注意:refs文件中的基因组参考序列和gtf文件以及几个vcf文件必须为同一版本,参考序列和相应的GTF文件必须为同一个网站的同一个版本,否则分析过程中会出现各种错误。很多文章推荐使用ensembl的版本,本文使用的是ucsc.hg19版本,因为之前ref文件和参考序列已经有了,只是增加了一个GTF文件,是从ucsc网站生成下载的,链接为:http://genome.ucsc.edu/cgi-bin/hgTables
分析流程:
-
01 INPUT 输入文件

-
02 之前用fastqc软件看过,adapter已经去掉了,这里直接开始align,Star Align

#创建一个变量star_length,值为read_length的值-1. let star_length=${envis.read_length}-1 #如果该star_length前缀的目录不存在,说明没有创建过索引,首先创建索引 if [ ! -d "/opt/ref/RNA/$star_length-1-pass-index" ]; then mkdir -p /opt/ref/RNA/$star_length-1-pass-index ${tools.STAR} --runMode genomeGenerate \ --runThreadN ${envis.threads} \ --genomeDir /opt/ref/RNA/$star_length-1-pass-index \ --genomeFastaFiles ${refs.hum} \ --sjdbGTFfile ${refs.gtf} \ --sjdbOverhang $star_length fi #使用创建的索引,执行align,为了节省空间设置了参数 --outSAMtype BAM Unsorted,也测试过sort类型,不能直接markdup ${tools.STAR} \ --genomeDir /opt/ref/RNA/$star_length-1-pass-index \ --runThreadN ${envis.threads} \ --readFilesIn ${data}/RNA/${sn}_R1.fastq.gz ${data}/RNA/${sn}_R2.fastq.gz \ --readFilesCommand zcat \ --sjdbOverhang $star_length \ --twopassMode Basic \ --outSAMtype BAM Unsorted \ --outFileNamePrefix ${result}/${sn}. -
03 Sort Bam:使用了sambamba替换了samtools

-
04-Mark duplicatate:使用了sambamba替换了gatk picard,重命名创建的索引与gatk命名一致。

-
05 SplitNCigarReads:将落在外显子上的reads分离出来,取出N错误碱基,去除内含子区域的reads。这一步太慢了,占用整个流程一半以上运行时间,不知道有没有办法提高速度。

-
06- 这一步添加sn样本编号等信息,前面sort如果使用samtools因为没有sn信息会报错。

-
07- BaseRecalibrator 碱基质量校正第一步

-
08- ApplyBQSR 应用碱基校正

-
09- 并行运行HaplotypeCaller

#清理拆分生成的interval文件夹:类似temp_0001_of_10,防止重复运行被上次的结果干扰 rm -rf ${result}/${sn}/temp_* #创建${sn}目录。 mkdir -p ${result}/${sn} #使用Gatk IntervalListTools拆分interval,拆分数量为${envis.scatter} ${tools.gatk} IntervalListTools \ --SCATTER_COUNT=${envis.scatter} \ --SUBDIVISION_MODE=BALANCING_WITHOUT_INTERVAL_SUBDIVISION_WITH_OVERFLOW \ --UNIQUE=true \ --SORT=true \ --INPUT=${refs.interval} \ --OUTPUT=${result}/${sn} #循环遍历生成的interval list文件,运行HaplotypeCaller index=1 for i in `ls ${result}/${sn}/*/scattered.interval_list`; do rm -f ${result}/${sn}_$index.vcf ${tools.gatk} HaplotypeCaller \ -L $i \ -R ${refs.hum} \ -I ${result}/${sn}_bqsr.bam \ -O ${result}/${sn}/${sn}_$index.vcf \ --dbsnp ${refs.dbsnp} \ -dont-use-soft-clipped-bases \ --standard-min-confidence-threshold-for-calling 20 & let index+=1 done #等待所有HaplotypeCaller运行结束 wait #用生成的vcf文件名拼接输入字段 vcfs= for z in `ls ${result}/${sn}/${sn}*.vcf`; do vcfs="-I $z $vcfs" done echo $vcfs #合并生成的vcf文件。 ${tools.gatk} MergeVcfs \ $vcfs \ -O ${result}/${sn}.vcf -
10-VariantFiltration:对于RNA-Seq,推荐使用硬过滤,不支持VQSR。

-
11-顺便使用fastqc看下QC结果:

-
12-最终分析结果:

运行结果
可以看到,GATK的工具一如既往的慢,HaplotypeCaller这一步通过拆分interval并行分析,最后合并vcf,速度从1个小时以上降到了9分钟。剩下的几步,SplitNCigarReads,BaseRecalibrator,ApplyBQSR都非常占用时间。也难怪市面上各种加速方案了。

















 354
354

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言










{kind=link}