










卫计委在2017年,2019年,2020年(还没有答案)提供标准数据用于肿瘤生信分析的室间质评。这样预知结果的数据自然是不能放过了,本文尝试参考GATK Best Practice:Somatic SNVs + Indels ,Cnvkit,Manta的pipeline来完成满分流程分析,也可以使用标准数据反向判断GATK Mutect2的实际准确度,算法优劣。
注:本文仅用于学习,距离真正的临床应用还有相当大距离,欢迎大佬批评指正
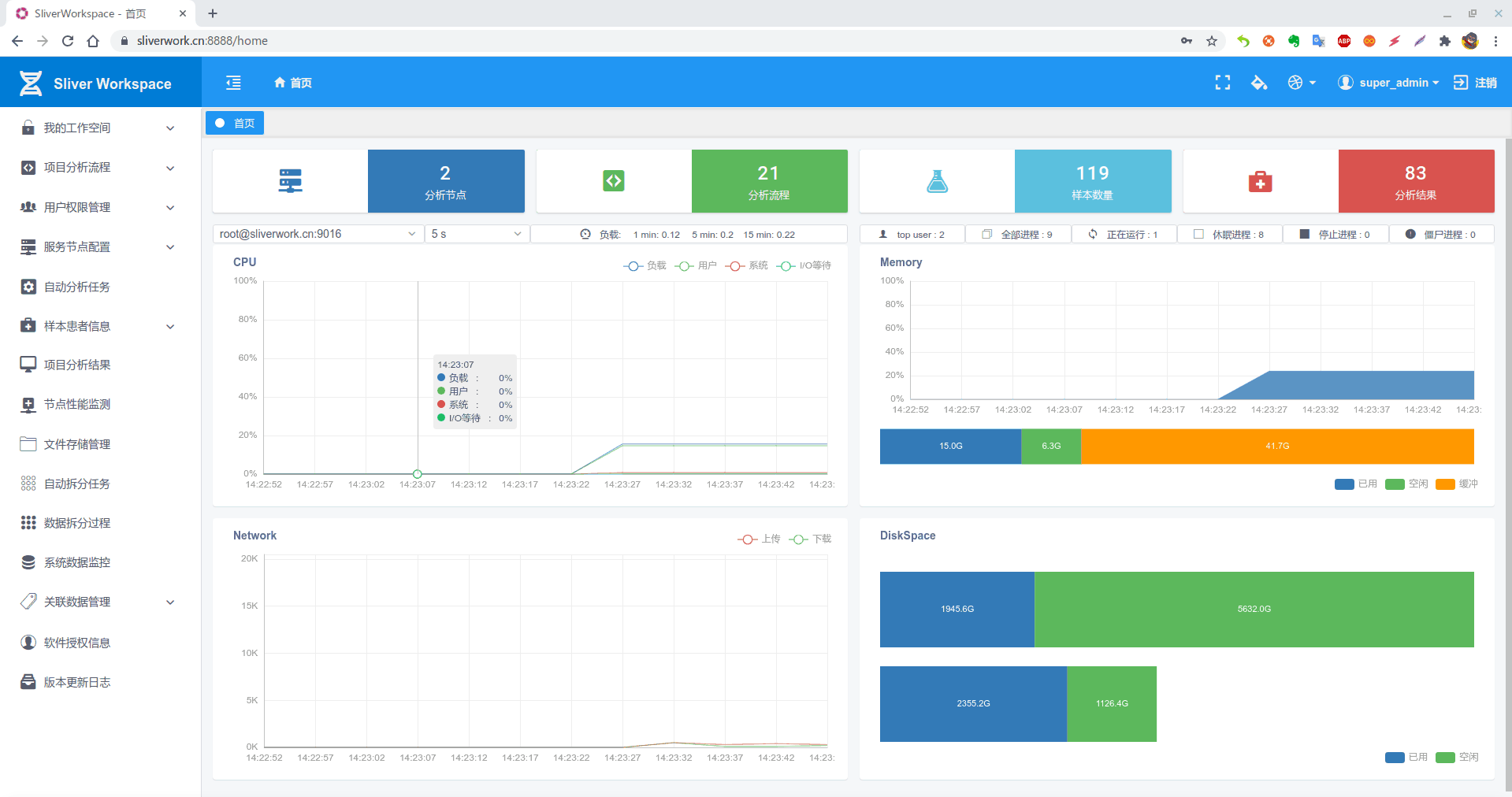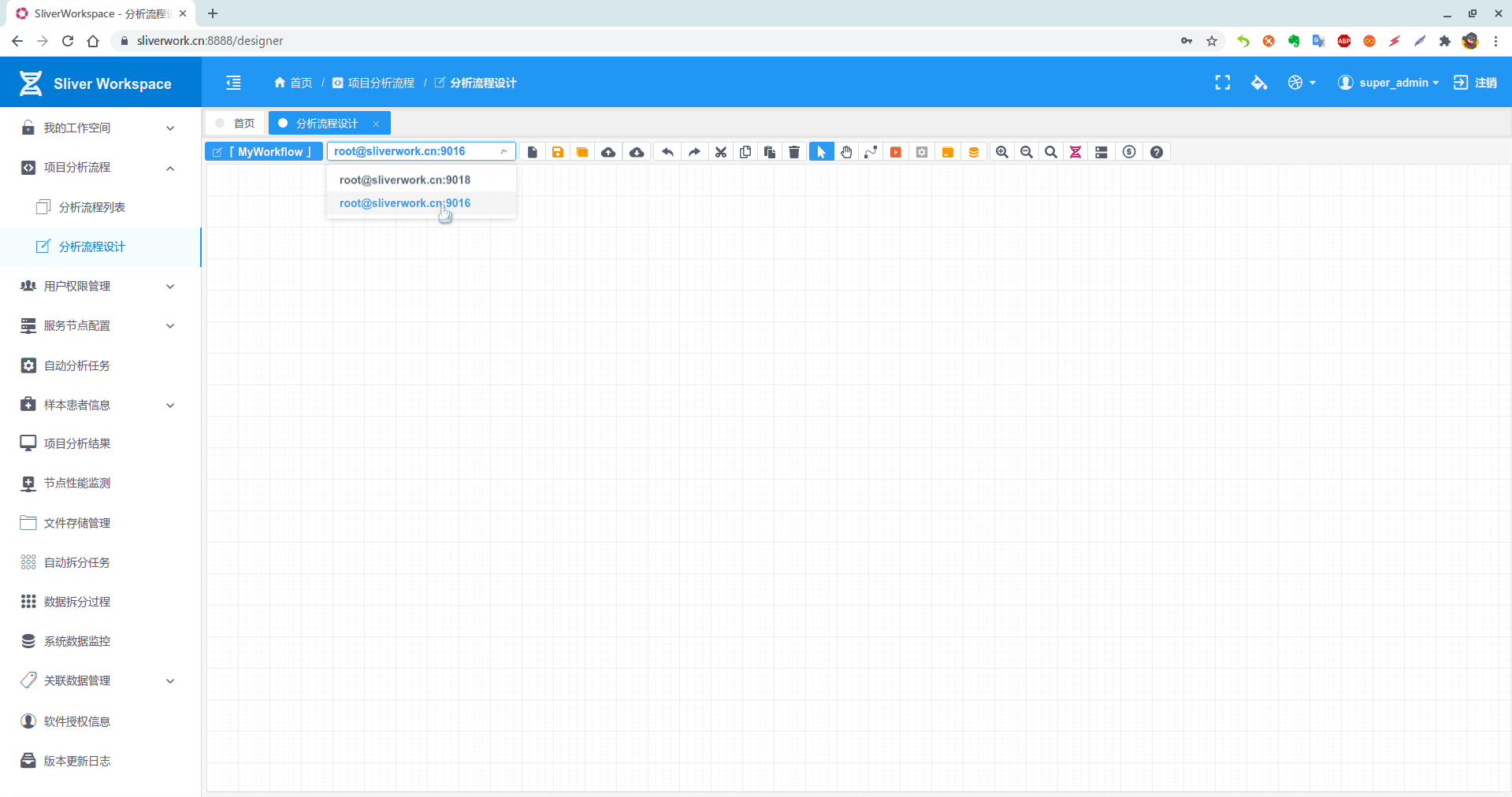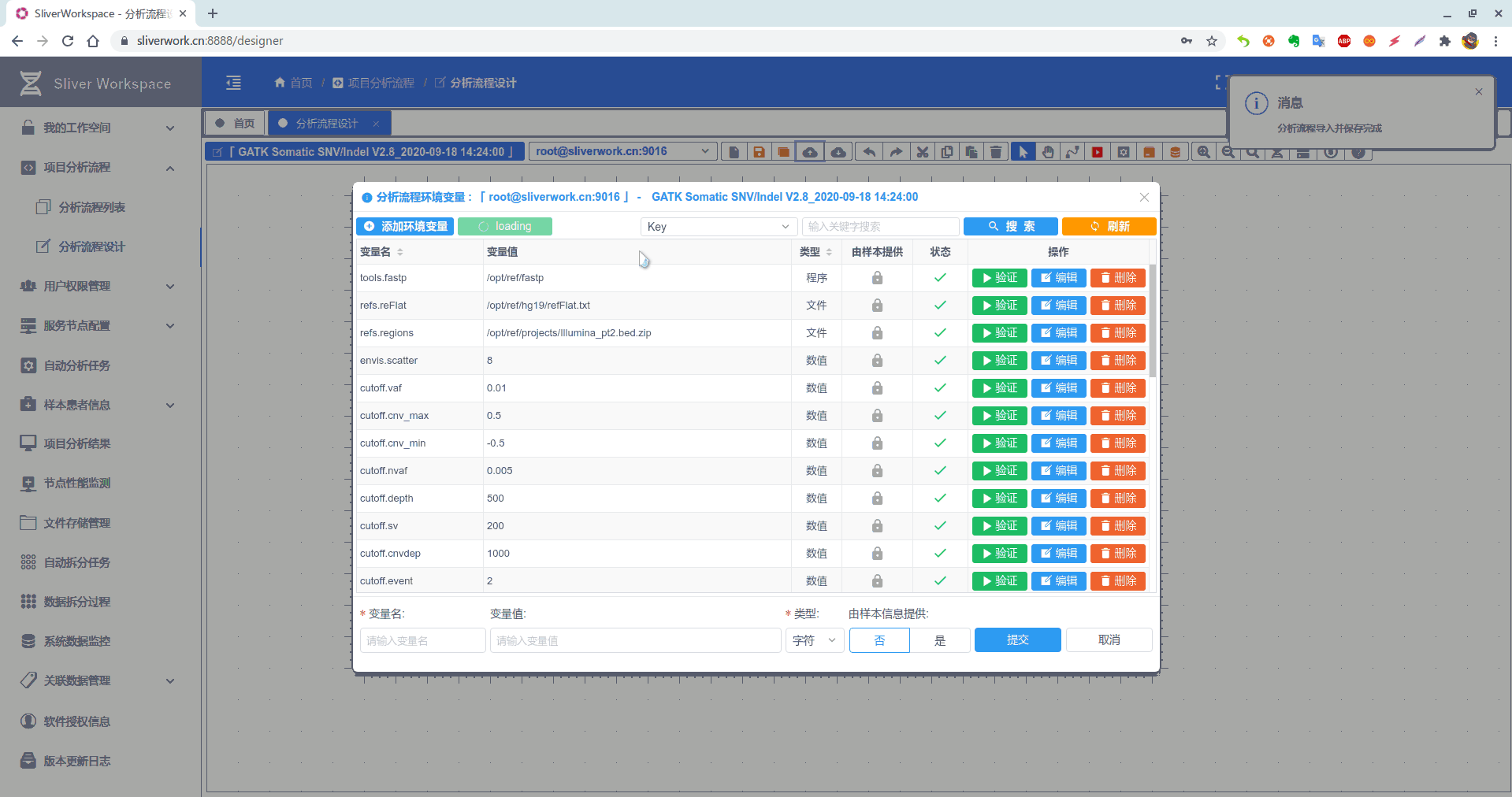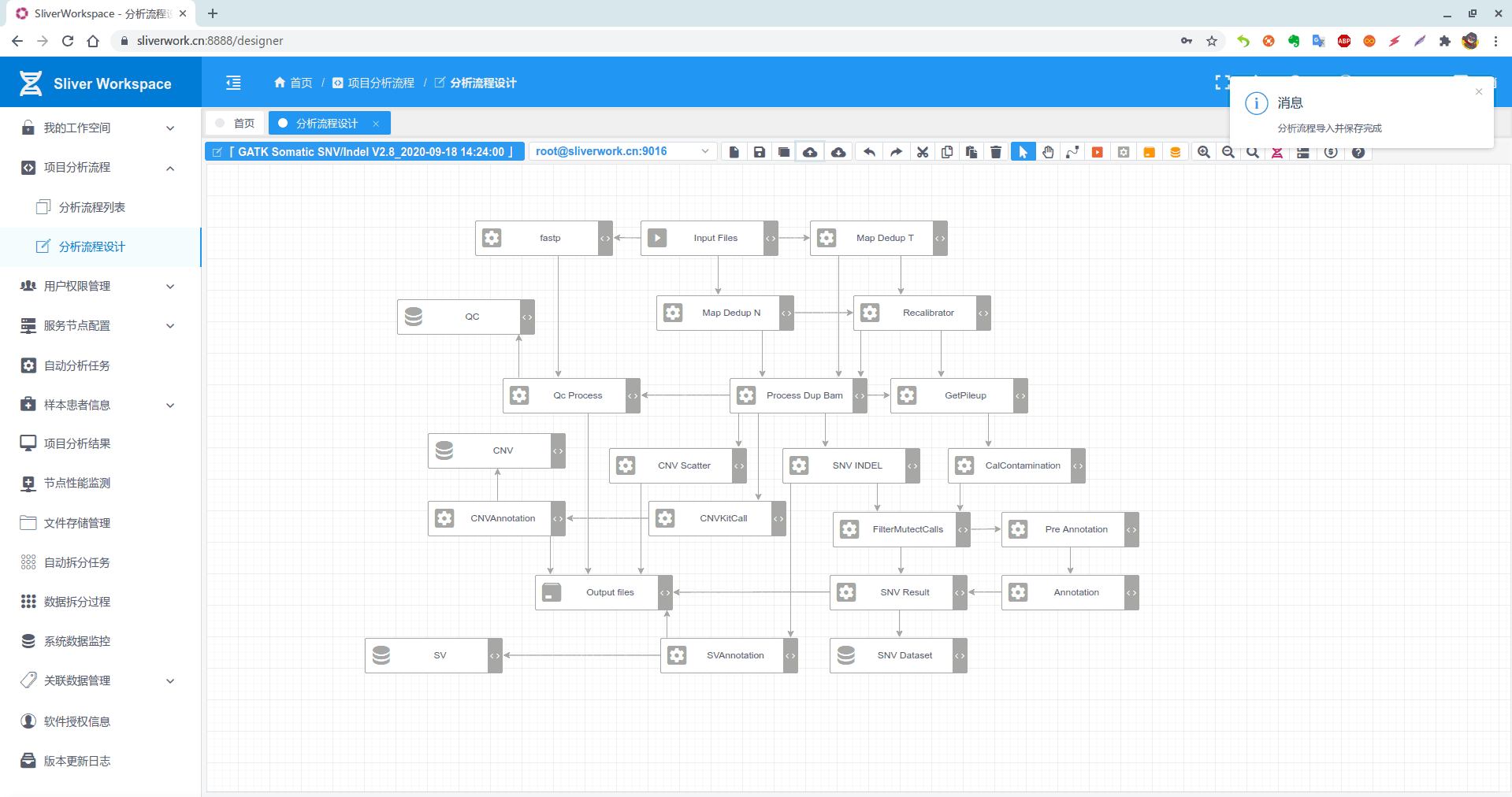
**1. 分析流程概览如下:

2. 本文用到的分析系统及分析流程文件
| 名称 (点击下载) | 备注 |
|---|---|
| SliverWorkspace 2.0.277363 | 提供运行控制平台/个人版 |
| FFPE SNV CNV SV V1.workflow | 分析流程文件,可以一键导入分析平台(点击查看操作) 当然可以参照图片中运行脚本,shell里运行,效果也是一样 |
| 最终结果过滤脚本(python2.7 )及编译版本 | Illumina_pt2.bed 等用到的bed,intelval等文件 SnvAnnotationFilter.py SNV过滤脚本 CnvAnnotationFilter.py CNV过滤脚本 SvAnnotationFilter.py SV 过滤脚本 QcProcessor.py 获取整体QC数据的脚本 report_template.docx 分析报告模板 |
| 分析结果(pipeline结果与标准答案) | result.zip pipeline结果与标准答案 |
3. 本文用到的原始文件,用fastqc查看质量状态是clean data,Q值均高于30,这里就不需要去接头和QC了。
百度网盘:链接: https://pan.baidu.com/s/1t9R14aQNP6Xk4tq1wFWJpg 提取码: u5yp
| 文件名(按照需要有调整) | 文件大小 | MD5 |
|---|---|---|
| B1701_R1.fq.gz | 4.85G | 07d3cdccee41dbb3adf5d2e04ab28e5b |
| B1701_R2.fq.gz | 4.77G | c2aa4a8ab784c77423e821b9f7fb00a7 |
| B1701NC_R1.fq.gz | 3.04G | 4fc21ad05f9ca8dc93d2749b8369891b |
| B1701NC_R2.fq.gz | 3.11G | bc64784f2591a27ceede1727136888b9 |
4. 本文用到的环境变量(目录/程序/文件/数值/字符)reference文件和数据库体积过于庞大请自行下载安装(如:ftp.broadinstitute.org/Annovar等等)
| 名称 | 数值 | 类型 |
|---|---|---|
| sn | 1701 | 字符 |
| data | /opt/data | 目录 |
| result | /opt/result | 目录 |
| tools.java | /opt/jdk1.8.0_162/bin/java | 程序 |
| tools.bgzip | /opt/tabix-0.2.6/bgzip | 程序 |
| tools.bwa | /opt/bwa/bwa | 程序 |
| tools.cnvkit | /usr/local/bin/cnvkit.py | 程序 |
| tools.fastqc | /opt/FastQC/fastqc | 程序 |
| tools.gatk | /opt/ref/gatk-4.1.3.0/gatk | 程序 |
| tools.manta | /opt/manta/dist/bin/configManta.py | 程序 |
| tools.samtools | /opt/samtools/samtools | 程序 |
| tools.tabix | /opt/tabix-0.2.6/tabix | 程序 |
| tools.annovar | /opt/ref/annovar/table_annovar.pl | 程序 |
| tools.convertor | /opt/ref/annovar/convert2annovar.pl | 程序 |
| cutoff.cnv_max | 0.5 | 数值 |
| cutoff.cnv_min | -0.5 | 数值 |
| cutoff.cnvdep | 1000 | 数值 |
| cutoff.depth | 500 | 数值 |
| cutoff.event | 2 | 数值 |
| cutoff.nvaf | 0.005 | 数值 |
| cutoff.sv | 200 | 数值 |
| cutoff.TLOD | 16 | 数值 |
| cutoff.vaf | 0.01 | 数值 |
| envis.scatter | 8 | 数值 |
| envis.threads | 32 | 数值 |
| envis.memory | 32G | 字符 |
| refs.1000G | /opt/ref/1000G_phase1.indels.hg19.vcf | 文件 |
| refs.af_only | /opt/ref/af-only-gnomad.raw.sites.hg19.vcf.gz | 文件 |
| refs.bed | /opt/ref/projects/Illumina_pt2.bed | 文件 |
| refs.interval | /opt/ref/projects/Illumina_pt2.interval_list | 文件 |
| refs.dbsnp | /opt/ref/dbsnp_138.hg19.vcf | 文件 |
| refs.dict | /opt/ref/ucsc.hg19.dict | 文件 |
| refs.gene | /opt/ref/hg19_refGene.txt | 文件 |
| refs.hum | /opt/ref/ucsc.hg19.fa | 文件 |
| refs.mills | /opt/ref/Mills_and_1000G_gold_standard.indels.hg19.vcf | 文件 |
| refs.small.exac | /opt/ref/small_exac_common_3_b37.vcf | 文件 |
5. 详细流程分析具体如下(不习惯图形软件的使用shell变量+脚本运行也是一样的)
GATK提供的标准流程从Normal/Tumor的fastq文件开始 map>reorder>sort>mark duplicate>recalibrator>apply BQSR
- 流程输入文件

分析流程输入文件,这里使用变量${sn}表示样本编号,对室间质评文件名做了调整。
-
Tumor 比对,管道操作给samtools,直接输出bam格式文件。

-
Reorder Bam
GATK文档中这样的
Not to be confused with SortSam which sorts a SAM or BAM file with a valid sequence dictionary, ReorderSam reorders reads in a SAM/BAM file to match the contig ordering in a provided reference file, as determined by exact name matching of contigs. Reads mapped to contigs absent in the new reference are dropped. Runs substantially faster if the input is an indexed BAM file.
这一步省略其实对最终结果影响不大。

-
Sort

-
MarkDuplicate 标记重复

-
重新校正碱基质量值第一步,BaseRecalibrator:计算所有需要重校正的reads和特征值,然后把这些信息输出为校准表文件

-
重新校正碱基质量值第二步,ApplyBQSR:用第一步得到的校准表文件,重新调整BAM文件中的碱基质量值,并使用这个新的质量值重新输出一个新的BAM文件。

-
Normal 数据对着Tumor的步骤重复一遍。序列比对

-
Normal Reorder

-
Normal Sort

-
Normal MarkDuplicate

- Normal BaseRecalibrator

-
Normal ApplyBQSR

-
Tumor GetPileupSummaries

-
Normal GetPileupSummaries

-
CalculateContamination

-
Mutect2

-
FilterMutectCalls 使用GATK提供的过滤器,过滤得到的突变

-
将过滤后的文件转换为Annovar注释所需要的格式

-
使用Annovar对结果注释

table_annovar.pl ${result}/${sn}.annovar \
/opt/ref/annovar/humandb \
-buildver hg19 \
-out ${result}/${sn} \
-remove \
-protocol refGene,avsnp150,esp6500siv2_all,1000g2015aug_all,clinvar_20190305,cosmic89_coding \
-operation g,f,f,f,f,f \
-nastring NAN
-
使用自己写的脚本对注释后的结果过滤,比如按照室间质评要求,过滤掉突变频率低于1%,测序深度低于500的突变。对GATK某些过滤器过滤掉的结果进行保留和排除,后面使用IGV进行人工筛选。最终输出的结果为,${sn}.result.SNV.xls(其实是个csv文件,扩展名改为.xls是便于使用excel打开,很多人都这么干)

-
使用Manta Call SV,第一步,生成分析脚本文件runWorkflow.py

-
运行runWorkflow.py获取SV突变结果

-
使用自己过滤的脚本文件,对Manta获取的SV过滤。如根据SOMATICSCORE分数过滤,根据hg19_refGene.txt提供文件,计算突变基因等等。

-
使用CnvKIt,获取CNV突变


-
使用CnvKit画图

-
使用py脚本文件,对CnvKit输出结果过滤。同样根据hg19_refGene.txt文件匹配基因,以及发生拷贝数变异的区域的外显子区域等。

-
获取整体QC数据,insertSize,depth等

- Samtools获取碱基测序深度

- Samtools flagstat统计比对信息

- 使用py脚本文件对以上获取的数据做统一处理,获取整体QC状态结果。

最终结果:

最终结果及各个命令/任务运行时间如下:

整个分析过程耗时 3h 58min 29s,比较耗时,这个版本为了逻辑清晰一些,多数任务都是串行运行,没有很好利用计算资源。
后续文章会对整个流程进行优化(主要是并行化),最终分析时间在1h 13min左右(提升约3倍)。
GATK 输出结果中SNV&INDEL的准确度问题,经过反复试验,不论如何设置过滤参数,最终的结果始终会有假阴性问题,这是GATK(4.1.3.0)中个别过滤器的问题,目前的补救措施是将部分GATK过滤器过滤掉的结果仍然包含在最终结果中,再使用IGV工具人工过滤(官方文档也是这么推荐的),判断该结果是否可靠。















 4850
4850

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言










{kind=link}