







桓峰基因的教程不但教您怎么使用,还会定期分析一些相关的文章,学会教程只是基础,但是如果把分析结果整合到文章里面才是目的,觉得我们这些教程还不错,并且您按照我们的教程分析出来不错的结果发了文章记得告知我们,并在文章中感谢一下我们哦!
公司英文名称:Kyoho Gene Technology (Beijing) Co.,Ltd.该文章使用桓峰基因公众号里面单细胞分析教程即可实现,有需要类似思路的老师可以联系我们!单细胞测序数据分析教程整理如下:
SCS【4】单细胞转录组数据可视化分析 (Seurat 4.0)
SCS【6】单细胞转录组之细胞类型自动注释 (SingleR)
SCS【7】单细胞转录组之轨迹分析 (Monocle 3) 聚类、分类和计数细胞
SCS【8】单细胞转录组之筛选标记基因 (Monocle 3)
SCS【9】单细胞转录组之构建细胞轨迹 (Monocle 3)
SCS【10】单细胞转录组之差异表达分析 (Monocle 3)
SCS【11】单细胞ATAC-seq 可视化分析 (Cicero)
SCS【12】单细胞转录组之评估不同单细胞亚群的分化潜能 (Cytotrace)
SCS【13】单细胞转录组之识别细胞对“基因集”的响应 (AUCell)
SCS【15】细胞交互:受体-配体及其相互作用的细胞通讯数据库 (CellPhoneDB)
SCS【16】从肿瘤单细胞RNA-Seq数据中推断拷贝数变化 (inferCNV)
前 言
Copynumber Karyotyping of Tumors(copyKAT)从高通量单细胞 RNA-seq 数据推断人类肿瘤的基因组拷贝数和亚克隆结构。单细胞转录组分析被广泛用于研究人类肿瘤。然而,在肿瘤微环境中区分正常细胞类型和恶性细胞以及分辨肿瘤内的克隆亚结构仍然具有挑战性。为了解决这些挑战,开发了一种称为非整倍体肿瘤拷贝数核型分析(CopyKAT)的综合贝叶斯分割方法,从高通量单细胞转录组测序(scRNA-seq)数据的读取深度估计平均基因组分辨率为5 Mb的基因组拷贝数谱。应用CopyKAT分析了21种肿瘤的46,501个单细胞,包括三阴性乳腺癌、胰导管腺癌、间变性甲状腺癌、浸润性导管癌和胶质母细胞瘤,准确(98%)将癌细胞与正常细胞类型区分开来。在三种乳腺肿瘤中,CopyKAT解决了肿瘤基因表达差异的克隆亚群,如KRAS,以及包括上皮到间质转变、DNA修复、凋亡和缺氧等特征。这些数据表明,CopyKAT可以帮助分析多种人类实体肿瘤中的scRNA-seq数据。

分析思路
在统计学上,CopyKAT将贝叶斯方法与层次聚类相结合,计算单个细胞的基因组拷贝数分布,并从高通量单细胞转录组数据中定义克隆子结构。首先,单细胞转录组数据的Unique Molecular ldentifier(UMI)的基因表达矩阵作为CopyKAT的输入,通过它们的基因组坐标对它们进行排序,并对基因的排列进行注释。之后,用Freeman-Tukey变换来稳定方差,然后采用多项式动态线性建模矫正单细胞UMI计数矩阵中的异常值。下一步是建立一个高可信度的正常二倍体细胞子集,用来推测正常二倍体细胞的拷贝数基线值。为此,研究人员将所有单细胞集中到几个小的亚群分类中,并使用高斯混合模型估算每个分类的方差。通过严格的分类标准,具有最小估计方差的聚类被定义为"标准的二倍体细胞"。为了检测染色体断点,他们整合泊松-伽玛模型和马尔可夫链蒙特卡罗迭代生成每个基因窗口的后验均值,然后应用Kolmogorov-Smirnov检验对均值无显著差异的相邻窗口进行合并,然后计算每个窗口的最终拷贝数值,以此作为跨越每个细胞中相邻染色体断点的所有基因的后验平均值。
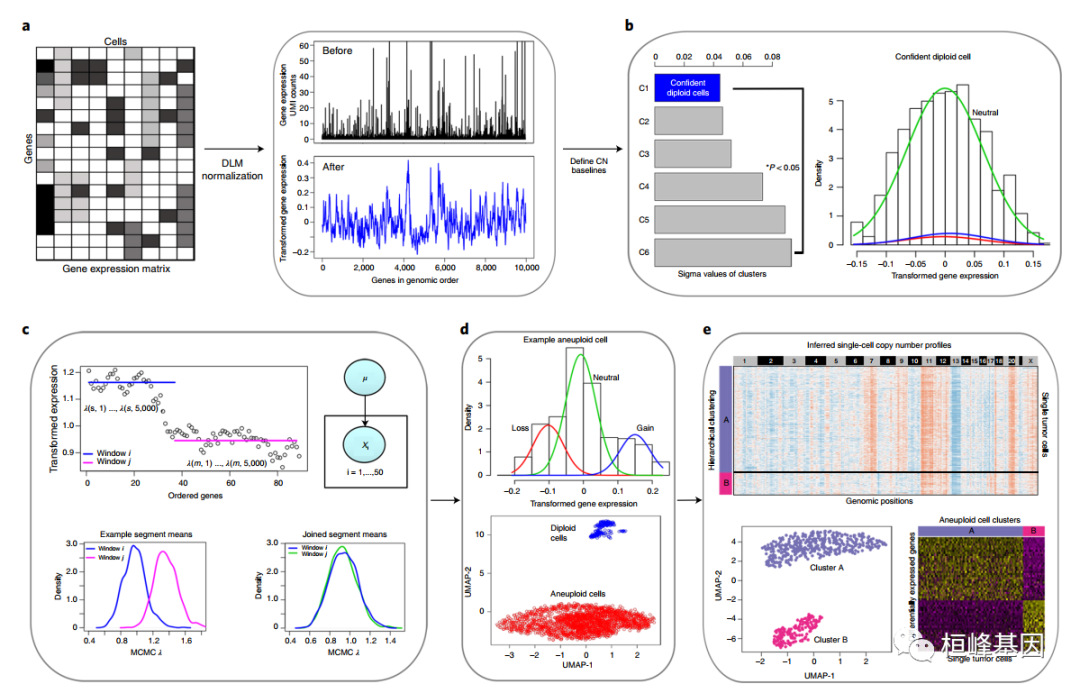
分析步骤
主要过程如下:
软件读取单细胞转录组数据的基因表达矩阵,根据坐标信息对基因进行排序注释。对原始计数矩阵执行对数 Freeman–Turkey 转换,以使用多项式 DLM 稳定方差和矫正异常值。
使用整合聚类和 GMM 方法定义高置信度的正常二倍体细胞子集,以推断正常二倍体细胞的拷贝数基线。
单个细胞中的相对基因表达值用于 MCMC 分割,并通过 KS 测试合并片段。
非整倍体肿瘤和正常细胞簇使用正常细胞富集和 GMM 分布测试进行分类。
通过聚类描绘肿瘤细胞的克隆亚结构,并使用亚克隆进行差异表达分析。
教程里面给出来5个步骤:
https://github.com/navinlabcode/copykat
Step 1: installation
Step 2: prepare the readcount input file
Step 3: running copykat
Step 4: navigate prediction results
Step 5: define subpopulations of aneuploid tumor cells
软件包安装
加载devtools从GitHub安装copykat
#Step 1: installation
library(devtools)
devtools::install_github("navinlabcode/copykat")数据读取
运行copykat需要准备的唯一输入文件是原始基因表达矩阵,其中基因id在行中,细胞名称在列中。基因id可以是基因符号,也可以是集合id。矩阵值通常是目前高通量单细胞RNA-seq数据的惟一分子标识符(UMI)计数。早期生成的scRNA-seq数据可以总结为TPM值或总读计数,这也有效。软件包例子提供一个从10X输出生成UMI计数矩阵的示例。我们这里直接使用Seurat软件包里面的例子 pbmc3k,读入代码如下:
# Step 2: prepare the readcount input file
library(copykat)
library(tidyverse)
library(Seurat)
library(SeuratData)
head(pbmc3k@meta.data)
exp.rawdata = as.matrix(pbmc3k@assays$RNA@data)
exp.rawdata
write.table(exp.rawdata, "exp.rawdata.txt", sep = "\t", quote = F)实例操作
现在已经准备了输入数据,原始UMI计数矩阵,准备运行copykat。为了过滤掉细胞,需要每个染色体上至少有5个基因来计算DNA拷贝数。可以调整参数 ngene。chr=1是为了保留尽可能多的细胞,但是我认为用至少5个基因代表一条染色体不是很严格。为了过滤掉基因,可以调整参数,只保留LOW表达的基因。DR到UP。细胞的DR组分。将默认设置为LOW.DR=0.05, UP.DR=0.2。我可以调低这些值,让更多的基因留在分析中确保LOW。DR比UP小。要求copykat每个片段至少取25个基因。我可以在每个bin内15-150个基因的范围内尝试。Cut是分割参数,取值范围为0 ~ 1。增加KS。切割降低灵敏度,即减少段/断点。通常在0.05-0.15的范围内工作。这里我通过设置n.cores = 4来进行并行计算。缺省值为1。通过设置 sam.name="test"给出了一个示例名称。一个令人挣扎的观察结果是,没有一种聚类方法能适合所有的数据集。在这个版本中,我为聚类添加了一个距离参数,包括“欧氏”距离和相关距离,即。1-“pearson”和“spearman”的相似性。一般来说,相关距离倾向于噪声数据,而欧氏距离倾向于具有较大CN段的数据。我添加了一个选项,将已知的普通单元格名称作为向量对象输入。默认值为NULL。我为只有非整倍体或二倍体细胞的细胞系数据添加了一个模式。通过cell.line="yes"设置此细胞行模式。组织样本的默认值是cell.line="no"。这种细胞模式使用从数据变化合成基线,不代表已发表的算法。这种细胞系模式不能保证成功或准确性。添加了一个选项输出seg文件,可以直接加载到IGV查看器进行可视化。默认为 output.seg="FALSE" 。如果需要seg文件,seg= "TRUE"。添加了一个额外的单细胞拷贝数结果图,使用基因按细胞基质。这是默认的plot.genes="TRUE"。基因名称标记在热图的底部。需要放大看小字体。由于请求较多,支持scRNA-seq。我还没有太多的老鼠数据集来测试这个方法。使用的方法与人类数据相同。唯一不同的是,结果是在基因空间而不是基因组空间中输出的。这意味着CNVs的位置是由基因名称标记的,而不是基因组位置。虽然非整倍体/二倍体预测被强制执行。运行包含10,000多个细胞的数据集可能需要一段时间。建议一次运行一个示例。将不同的样品组合在一起会产生样品之间的批处理效应。此步骤完成后,copykat自动将计算出的拷贝数矩阵、热图和肿瘤/正常预测结果保存到工作目录中。以下是两个例子,现在运行代码:
# Step 3: running copykat
library(copykat)
copykat.test <- copykat(rawmat = exp.rawdata, id.type = "S", ngene.chr = 5, win.size = 25,
KS.cut = 0.1, sam.name = "test", distance = "euclidean", norm.cell.names = "",
output.seg = "FLASE", plot.genes = "TRUE", genome = "hg20", n.cores = 1)
pred.test <- data.frame(copykat.test$prediction)
pred.test <- pred.test[-which(pred.test$copykat.pred == "not.defined"), ] ##remove undefined cells
CNA.test <- data.frame(copykat.test$CNAmat)现在看看预测结果。预测的非整倍体细胞被推断为肿瘤细胞;二倍体细胞是基质正常细胞。这个版本放回所有过滤过的细胞,并将它们标记为“未定义”的细胞。CNV矩阵的前3列是基因组坐标。行是基因组顺序的220KB bin。并输出以基因名称为索引的cnv和染色体名称、起始和结束位置、染色带等信息。小鼠结果仅输出细胞空间中基因名称的CNV结果。Copykat生成一个估计拷贝数的热图图。行是细胞;列是按基因组顺序排列的220kb bin。
# Step 4: navigate prediction results
head(pred.test)
head(CNA.test[, 1:5])
my_palette <- colorRampPalette(rev(RColorBrewer::brewer.pal(n = 3, name = "RdBu")))(n = 999)
chr <- as.numeric(CNA.test$chrom)%%2 + 1
rbPal1 <- colorRampPalette(c("black", "grey"))
CHR <- rbPal1(2)[as.numeric(chr)]
chr1 <- cbind(CHR, CHR)
rbPal5 <- colorRampPalette(RColorBrewer::brewer.pal(n = 8, name = "Dark2")[2:1])
com.preN <- pred.test$copykat.pred
pred <- rbPal5(2)[as.numeric(factor(com.preN))]
cells <- rbind(pred, pred)
col_breaks = c(seq(-1, -0.4, length = 50), seq(-0.4, -0.2, length = 150), seq(-0.2,
0.2, length = 600), seq(0.2, 0.4, length = 150), seq(0.4, 1, length = 50))
png("navigate_prediction_results.png", width = 1400, height = 1200)
heatmap.3(t(CNA.test[, 4:ncol(CNA.test)]), dendrogram = "r", distfun = function(x) parallelDist::parDist(x,
threads = 4, method = "euclidean"), hclustfun = function(x) hclust(x, method = "ward.D2"),
ColSideColors = chr1, RowSideColors = cells, Colv = NA, Rowv = TRUE, notecol = "black",
col = my_palette, breaks = col_breaks, key = TRUE, keysize = 1, density.info = "none",
trace = "none", cexRow = 0.1, cexCol = 0.1, cex.main = 1, cex.lab = 0.1, symm = F,
symkey = F, symbreaks = T, cex = 1, cex.main = 4, margins = c(10, 10))
legend("topright", paste("pred.", names(table(com.preN)), sep = ""), pch = 15, col = RColorBrewer::brewer.pal(n = 8,
name = "Dark2")[2:1], cex = 0.6, bty = "n")
dev.off()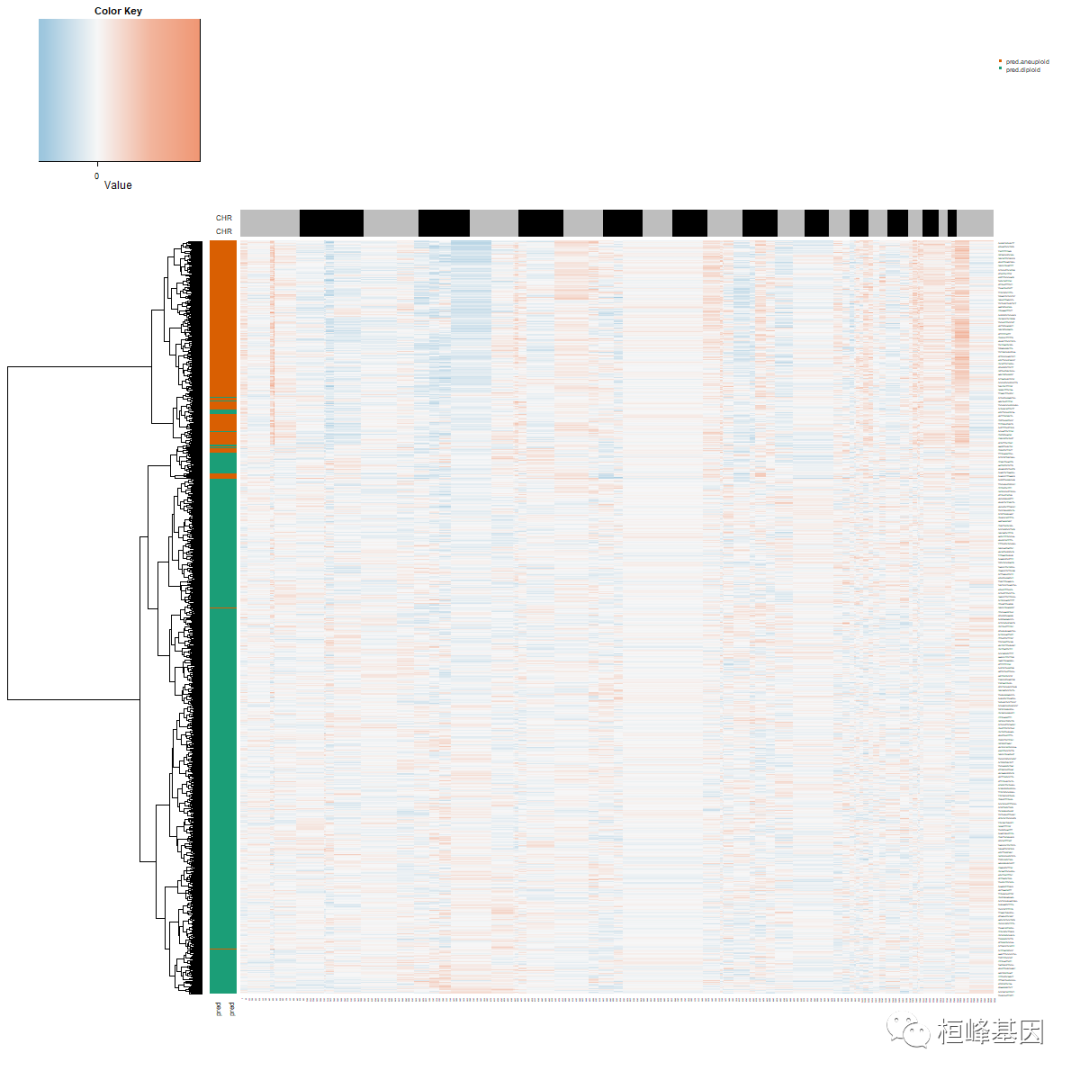
观察到了二倍体和非整倍体细胞。下一步是提取非整倍体肿瘤中被认为是肿瘤细胞的非整倍体细胞,定义单个肿瘤细胞的两个拷贝数亚群。
# Step 5: define subpopulations of aneuploid tumor cells
tumor.cells <- pred.test$cell.names[which(pred.test$copykat.pred == "aneuploid")]
tumor.mat <- CNA.test[, which(colnames(CNA.test) %in% tumor.cells)]
hcc <- hclust(parallelDist::parDist(t(tumor.mat), threads = 4, method = "euclidean"),
method = "ward.D2")
hc.umap <- cutree(hcc, 2)
rbPal6 <- colorRampPalette(RColorBrewer::brewer.pal(n = 8, name = "Dark2")[3:4])
subpop <- rbPal6(2)[as.numeric(factor(hc.umap))]
cells <- rbind(subpop, subpop)
png("subpopulations_aneuploid_tumor_cells.png", width = 1200, height = 1200)
heatmap.3(t(tumor.mat), dendrogram = "r", distfun = function(x) parallelDist::parDist(x,
threads = 4, method = "euclidean"), hclustfun = function(x) hclust(x, method = "ward.D2"),
ColSideColors = chr1, RowSideColors = cells, Colv = NA, Rowv = TRUE, notecol = "black",
col = my_palette, breaks = col_breaks, key = TRUE, keysize = 1, density.info = "none",
trace = "none", cexRow = 0.1, cexCol = 0.1, cex.main = 1, cex.lab = 0.1, symm = F,
symkey = F, symbreaks = T, cex = 1, cex.main = 4, margins = c(10, 10))
legend("topright", c("c1", "c2"), pch = 15, col = RColorBrewer::brewer.pal(n = 8,
name = "Dark2")[3:4], cex = 0.9, bty = "n")
dev.off()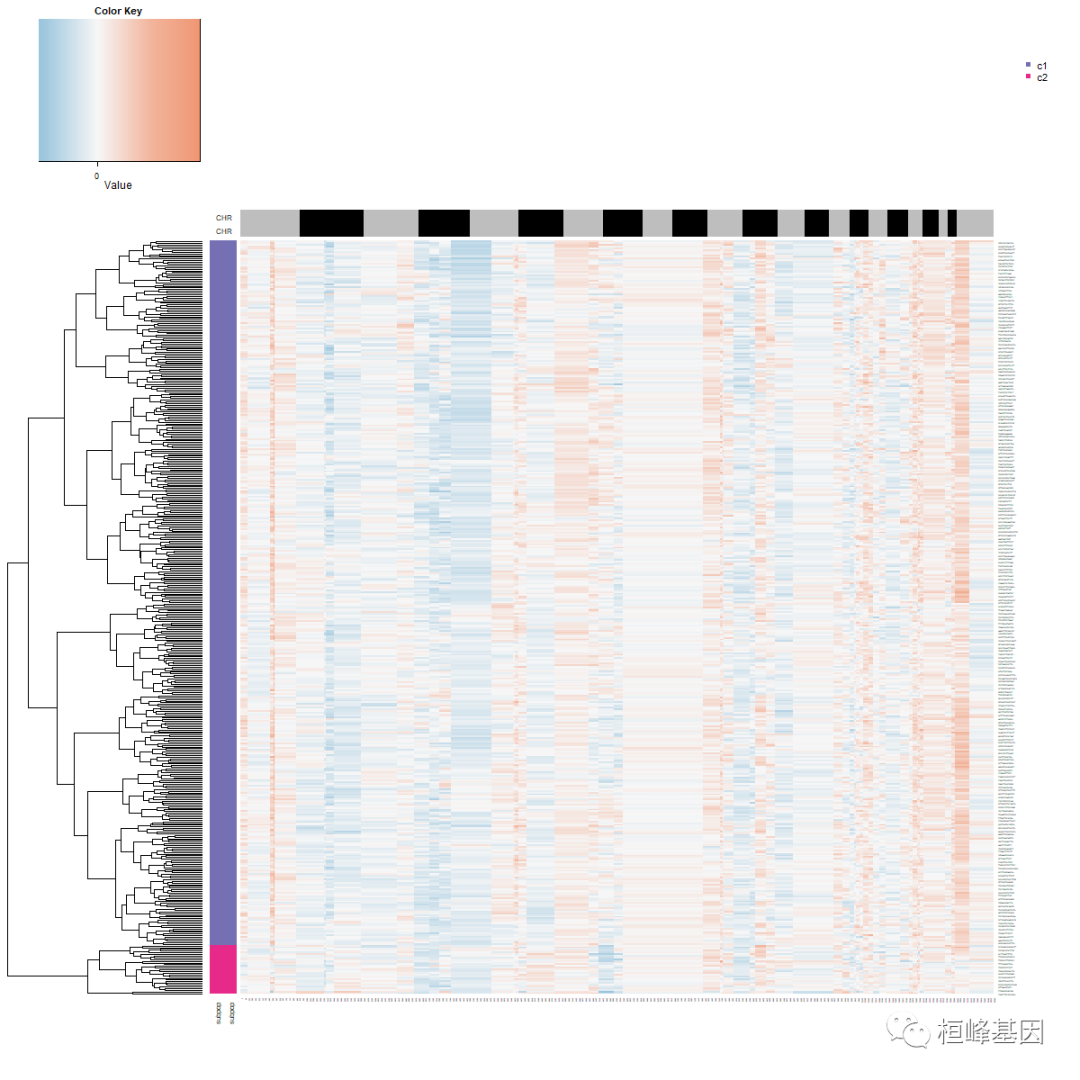
我们这期主要介绍从肿瘤单细胞RNA-Seq数据中推断拷贝数变异可以比较之前使用inferCNV的结果进行比较,这个copyKAT优势在于不需要有正常的细胞做对照就可以将肿瘤细胞挑选出来。目前单细胞测序的费用也在降低,单细胞系列可算是目前的测序神器,有这方面需求的老师,联系桓峰基因,提供最高端的科研服务!
桓峰基因,铸造成功的您!
未来桓峰基因公众号将不间断的推出单细胞系列生信分析教程,
敬请期待!!
有想进生信交流群的老师可以扫最后一个二维码加微信,备注“单位+姓名+目的”,有些想发广告的就免打扰吧,还得费力气把你踢出去!

References:
Gao, R., Bai, S., Henderson, Y. C., Lin, Y., Schalck, A., Yan, Y., Kumar, T., Hu, M., Sei, E., Davis, A., Wang, F., Shaitelman, S. F., Wang, J. R., Chen, K., Moulder, S., Lai, S. Y. & Navin, N. E. (2021). Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat Biotechnol. doi:10.1038/s41587-020-00795-2.

















 2265
2265

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









