







 超级会员免费看
超级会员免费看
 本文介绍了如何使用pymol命令行和spyrmsd库来计算分子或蛋白质的RMSD,以及RMSD在结构比较中的作用。详细步骤包括pymol中的python脚本应用和spyrmsd库的安装与使用,并提到了RMSE、MSE和MAE的区别。
本文介绍了如何使用pymol命令行和spyrmsd库来计算分子或蛋白质的RMSD,以及RMSD在结构比较中的作用。详细步骤包括pymol中的python脚本应用和spyrmsd库的安装与使用,并提到了RMSE、MSE和MAE的区别。
参考:https://zhuanlan.zhihu.com/p/347743101
https://www.codenong.com/cs106148400/
RMSD
单位是埃 RMSD,root-mean-square deviation,也就是均方根偏差。 原子位置的均方根偏差是叠加蛋白质的原子(通常是骨架原子)之间的平均距离的量度。注意,RMSD计算可以应用于其他非蛋白质分子,如小的有机分子。在球状蛋白质构象的研究中,通常在刚体进行完叠加后通过计算Cα原子坐标之间的RMSD来表征三维结构的相似性。 等式:
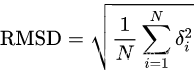
通常,RMSD用作两种或更多种蛋白质结构之间相似性的定量测量,通常越低越好。
命令行
#rank1, 4o75_ligand是两个分子
align rank1, 4o75_ligand

另外方法:
pymol 加载python计算代码:

 订阅专栏 解锁全文
订阅专栏 解锁全文



















 9960
9960

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











