







AMBER分子动力学模拟之结果分析(MMGB/PBSA)-- HIV蛋白酶-抑制剂复合物(4)
结合自由能计算
我们首先计算焙变,用到的是pbsa和gbsa方法。我们需要一下文件
三个top文件,pro.prmtop lig.prmtop com.prmtop;输入文件MM_GBSA.in;将要进行运算的轨迹文件md2.crd;执行文件run.sh;
MMGBSA
vim MM_GBSA.in
## 所用轨迹的参数
&general
startframe = 1, ## 帧的开始
endframe = 400, ## 帧的结束
interval=40, ## 帧间隔
use_sander =1, #
netcdf=1, ##轨迹是压缩格式吗
keep_files=0, ## 是否保存中间文件
debug_printlevel = 0,
verbose = 1,
entropy = 0 ## 是否计算熵
/
### 计算GBSA的参数
&gb
igb = 2, # GB类型
saltcon = 0, # 带电吗
ifant = 0, #
molsurf = 0,
surften = 0.005,
surfoff = 0
/
## 是否做残基分解
&decomp
idecomp=1,
dec_verbose=0
/
## 是否用nmode计算熵变
#&nmode
#nmstartframe =1,
#nmendframe =10,
#nminterval =1,
#dielc = 1,
#maxcyc = 500000,
#drms = 0.001,
#nmode_igb =0
#/
vim run.sh
单核计算
python MMPBSA.py -O -i MM_GBSA.in -o MM_GBSA.dat -eo MM_GBSA.csv -do MM_GBSA_DECOMP.dat -deo MM_GBSA_DECOMP.csv -cp com.top -rp pro.top -lp ../top/lig.top -y ../md/md2.crd> MM_PBGBSA.log
并行计算
mpirun -np2 MMPBSA.py.MPI -O -i MM_GBSA.in -o MM_GBSA.dat -eo MM_GBSA.csv -do MM_GBSA_DECOMP.dat -deo MM_GBSA_DECOMP.csv -cp com.top -rp pro.top -lp ../top/lig.top -y ../md/md2.crd> MM_PBGBSA.log
参数说明
-np 2 2个core并行
-i MM_GBSA.in input文件
-o MM_GBSA.dat 结果文件
-eo MM_GBSA.csv 详细结果文件
-do MM_GBSA_DECOMP.dat 残基分解的结果文件(总的)
-deo MM_GBSA_DECOMP.csv 残基分解的详细结果文件(每一帧)
-cp com.top 复合物的 top
-rp pro.top 文件受体的 top
-lp lig.top 文件配体的 top
-y …/md2/md2.crd 文件轨迹文件
MMPBSA
## 所用轨迹的参数
&general
startframe = 1, ## 帧的开始
endframe = 400, ## 帧的结束
interval=40, ## 帧间隔
use_sander =1, #
netcdf=1, ##轨迹是压缩格式吗
keep_files=0, ## 是否保存中间文件
debug_printlevel = 0,
verbose = 1,
entropy = 0 ## 是否计算熵
/
&pb
indi=1,
exdi=80.0
inp=1
cavity_offset=0.92,
scale=2.0,
istrng=0.1,
linit=1000,
prbrad=1.4,
radiopt=0
/
&decomp
idecomp=1,
dec_verbose=0
/
## 是否用nmode计算熵变
#&nmode
#nmstartframe =1,
#nmendframe =10,
#nminterval =1,
#dielc = 1,
#maxcyc = 500000,
#drms = 0.001,
#nmode_igb =0
#/
vim run.sh
MMPBSA.py -O -i MM_PBSA.in -o MM_PBSA.dat -eo MM_PBSA.csv -do MM_PBSA_DECOMP.dat -deo MM_PBSA_DECOMP.csv -cp com.top -rp pro.top -lp ../top/lig.top -y ../md/md2.crd> MM_PBGBSA.log
mpirun -np2 MMPBSA.py.MPI -O -i MM_PBSA.in -o MM_GBSA.dat -eo MM_PBSA.csv -do MM_PBSA_DECOMP.dat -deo MM_PBSA_DECOMP.csv -cp com.top -rp pro.top -lp ../top/lig.top -y ../md/md2.crd> MM_PBGBSA.log
结果分析
运行结果MMGBSA.dat,详细的每一帧的结果在MMGBSA.csv
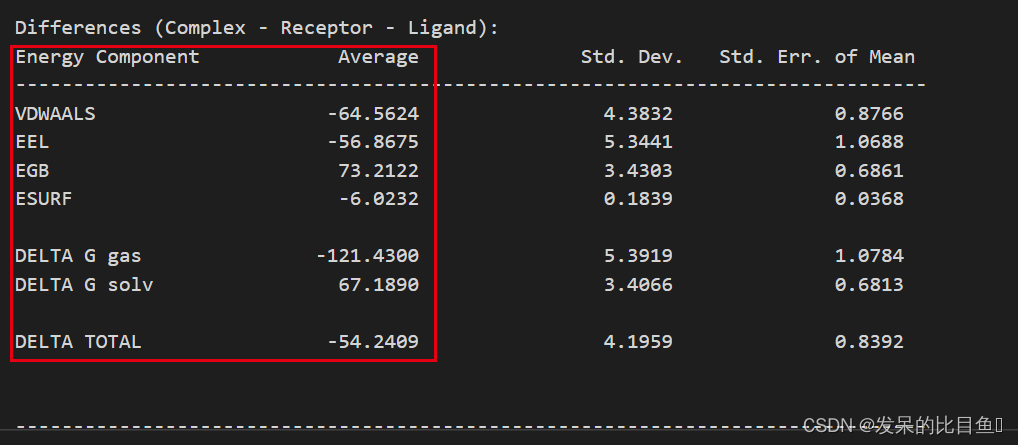
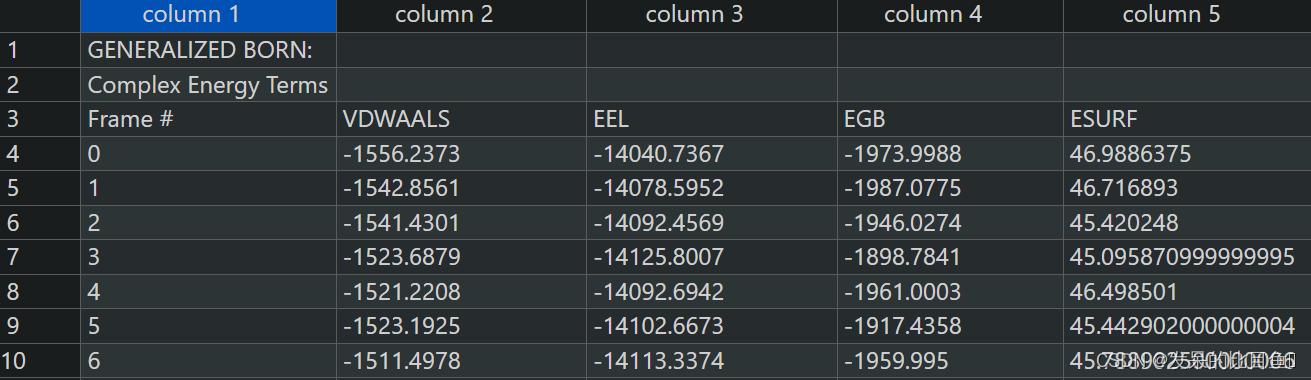
残基分解的结果MM_GBSA_DECOMPdat,详细的每一帧的结果在MM_GBSA_DECOMP.csv
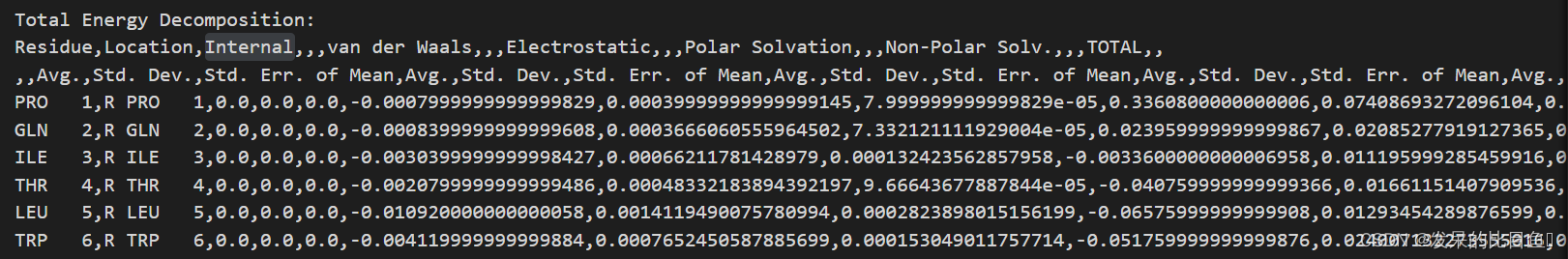
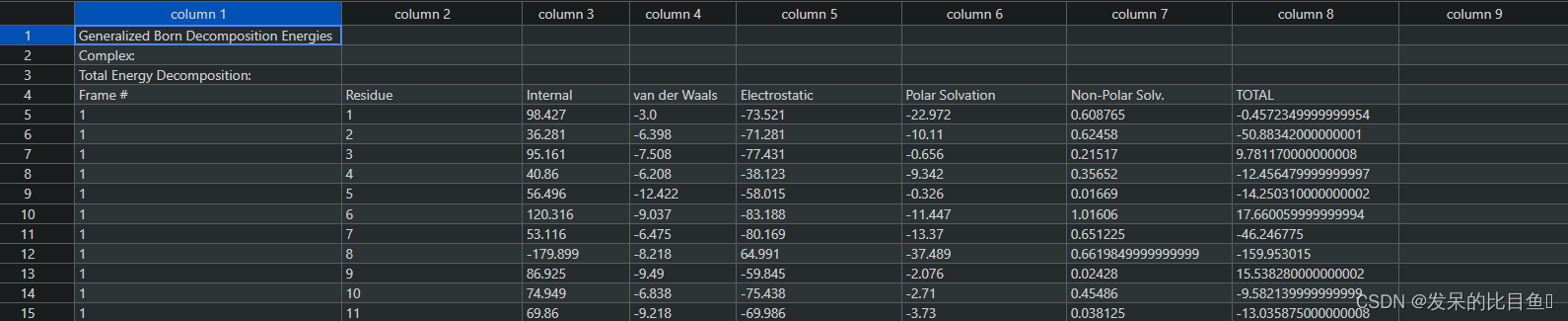
















 2802
2802











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











