









01 blast原理介绍
The Basic Local Alignment Search Tool (BLAST) finds regions of local similarity between sequences. The program compares nucleotide or protein sequences to sequence databases and calculates the statistical significance of matches. BLAST can be used to infer functional and evolutionary relationships between sequences as well as help identify members of gene families.
基本局部比对搜索工具(BLAST)用于找到序列之间的局部相似区域。该程序将核苷酸或蛋白质序列与序列数据库进行比较,并计算匹配的统计学意义。BLAST可用于推断序列之间的功能和进化关系,以及帮助识别基因家族的成员。
序列比对是指利用计算机程序比较核酸或蛋白质序列之间相似性,找出两个或多个序列之间的相同区域或差异位点。根据分子生物学中心法则,DNA是遗传信息携带者,而蛋白质则是功能分子。不同物种之所以千姿百态、各不相同,其内在原因是它们的基因组不同,或者更确切地说,是它们的DNA序列及其编码所得的蛋白质不同。
1.1 序列同源性与相似性
序列比对经常用来判断所比对的两个序列是否为同源序列。必须指出,相似性(Similarity)和同源性(Homology)是两个完全不同的概念。根据达尔文进化论学说,地球上现有物种,不论是动物、植物,或者是微生物,可以追溯到一个共同祖先。由于地球环境不断变化,祖先物种在演化过程中发生分化,形成新物种,以适应变化后的新环境。演化过程中,不同物种的基因组核酸序列及其编码的蛋白质序列均发生不同程度的突变,但依然保持一定的相似性。
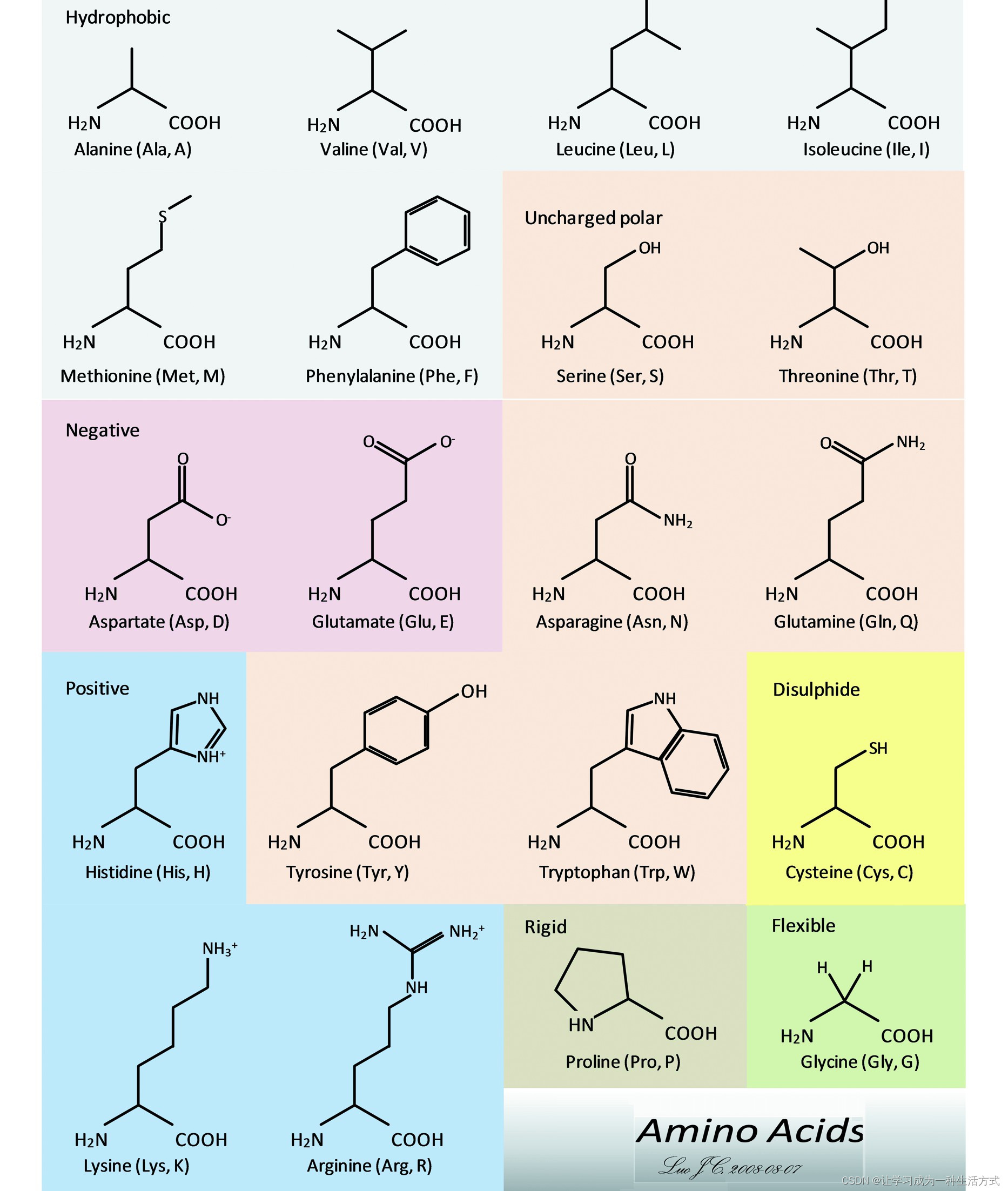
序列相似性概念在核酸和蛋白质序列中有所不同。核酸序列的相似性高低,是指通过序列比对所得结果中相同核苷酸残基所占比例,通常用百分比表示。而蛋白质序列比对结果中,除了用相同氨基酸残基所占比例作为相似性指标外,也经常用相同氨基酸加上相似氨基酸作为相似性指标。所谓相似氨基酸,是指侧链基团理化性质相似的一对氨基酸,如疏水氨基酸亮氨酸(Leu, L)和异亮氨酸(Ile, I),极性氨基酸丝氨酸(Ser, S)和苏氨酸(Thu, T)、带负电的氨基酸门冬氨酸(Asp, D和谷氨酸(Glu, E)、带苯环的氨基酸苯丙氨酸(Phe, F)和酪氨酸(Tyr, Y)等。
不论是核酸序列还是蛋白质序列,序列相似性是指相同和相似残基所占全长序列的比例,比例越高,相似性越高。而序列同源性是指所比较的两个序列是否具有共同的祖先序列。显然,序列同源性只有是非之别,没有高低之分,所谓“具有50%同源性”,或“高度同源”等说法,都是错误的。值得一提的是,相似性和同源性的概念之所以容易混淆,是因为两者之间关系密切。一般说来,同源序列特别是亲缘关系较近的序列,相似性通常较高;反之,相似性较高的两条序列,很有可能具有共同祖先。也就是说,序列相似性的高低经常用来推断其是否同源。
同源序列通常分为直系同源(Ortholog)和并系同源(Paralog)两类。以血红蛋白为例,小鼠和大鼠两个不同物种alpha血红蛋白在它们共同祖先中已经存在。随着物种分化,小鼠和大鼠共同祖先分化为两个物种,所形成的新物种通过遗传机制获得祖先物种基因组中alpha珠蛋白基因。因此,小鼠和大鼠两个不同物种中alpha血红蛋白称为直系同源蛋白,其编码基因则为直系同源基因。而小鼠中alpha珠蛋白基因在物种形成后,由基因复制产生了两个基因,编码两个alpha血红蛋白,即alpha1和alpha2。小鼠血红蛋白alpha1和alpha2则称为并系同源(有时也译作旁系同源)蛋白,其编码基因则称为并系同源基因。
1.2 blast算法矩阵与罚分
序列相似性数据库搜索软件Basic Local Alignment Search Tool(BLAST)则采用启发式算法。BLAST通常用于搜索某个蛋白质或核酸序列数据库中与检测序列具有一定相似性的靶标序列。
BLAST算法大体分为以下三步。首先,将检测序列按一定字长(Word Size)拆分成种子(Seed)序列,并按给定计分矩阵和设定阈值,找到与种子序列相似性较高的近邻(Neighbor)序列。接着,逐个找到各近邻序列在数据库中匹配序列,并按分值增加原则向两边延伸,得到高分对(High Scoring Pair)。将所得主对角线方向距离较近的高分对连接起来,并用Smith-Waterman方法进行比对。最后,对搜索到的靶标序列进行统计检验,输出期望值(Expect Value)低于设定阈值的靶标序列,即搜索结果。BLAST也可用于双序列比对,只要把所要搜索的数据库设定为另一个序列。显然,由于所采用的比对策略完全不同,基于Smith-Waterman动态规划算法的比对结果和基于BLAST启发式算法的比对结果不一定相同,某些情况下差别很大。
无论整体比对还是局部比对,无论采用动态规划算法还是采用启发式算法,都离不开计分矩阵和空位罚分。所谓计分矩阵,是指比对过程中相同或不同核苷酸或氨基酸之间的匹配或错配分值。例如,核酸序列比对时通常匹配分值为正值,而错配分值为负值。蛋白质序列比对时,匹配分值为正值,而错配分值则与氨基酸性质有关,性质不同的氨基酸之间的错配分值为负值,而性质相似的氨基酸之间的分值有可能为正值。不同计分矩阵具有不同匹配分值和错配分值。核酸序列比对中常用的DNAfull计分矩阵,以及蛋白质序列比对中常用的BLOSUM62和PAM250计分矩阵。显然,计分矩阵不同,比对结果很可能不同。这一点,实际使用时经常被忽略。
核苷酸计分矩阵DNAfull
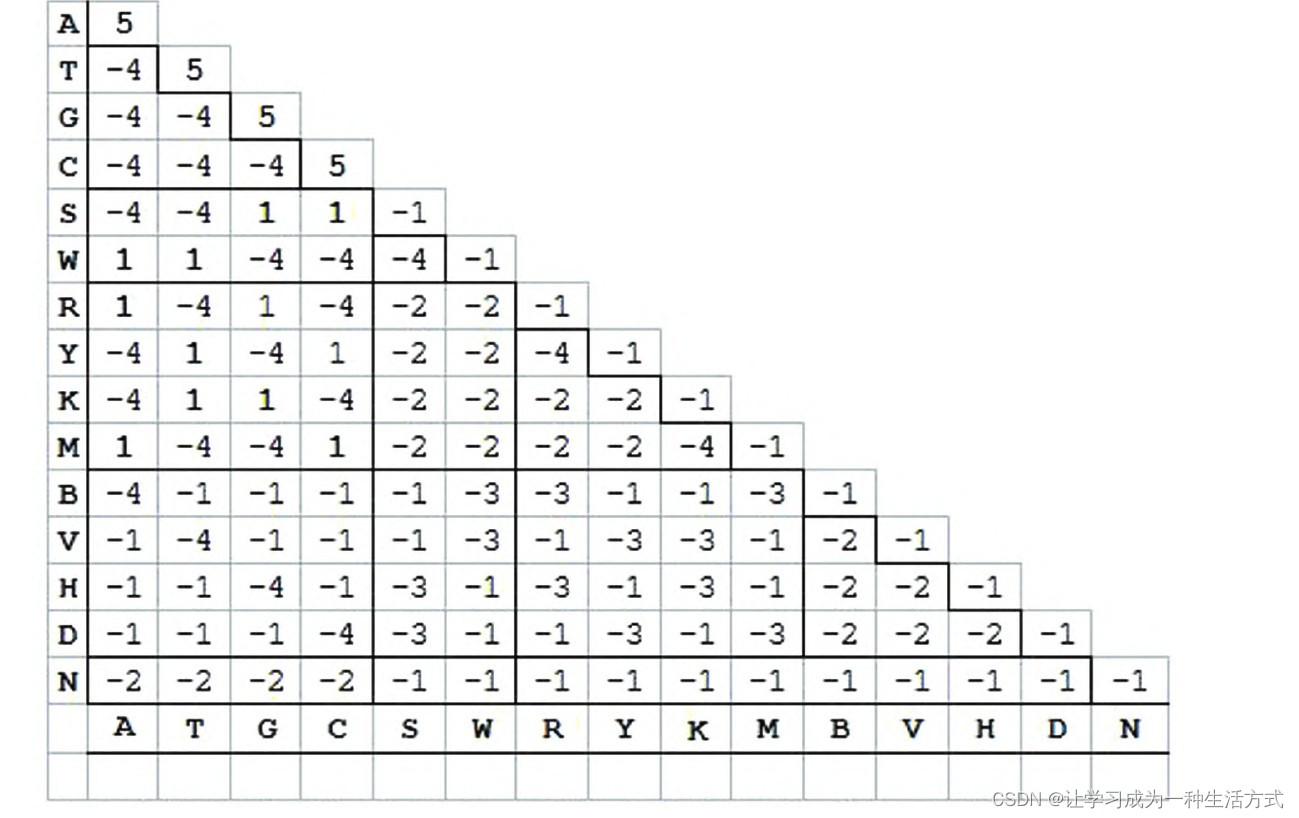
相似性计分矩阵BLOSUM62

序列比对的过程就是利用一定算法或策略,确定是否插入空位、何处插入空位、插入几个空位。这一看似简单的过程,在计算机领域实际上是个难度极大的复杂计算问题,而其背后的生物学机制则更加复杂。所谓空位罚分,是指比对过程中在适当位置插入空位,使比对总分值更高、比对结果更好。实际比对时,程序通常给定默认值,而用户可以根据具体情况进行调整。空位罚分大小设置通常采用经验值,起始空位罚分较大,而延伸空位罚分较小。所谓起始空位,是指插入的第一个空位,而延伸空位则是当插入多个空位时,第二个空位开始的其它空位。此外,当两个长度差别较大的序列进行整体比对时,往往需要考虑是否对末端空位也进行罚分。
02 参考
https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ #ncbi地址03 安装
wget -c https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ncbi-blast-2.14.0+-x64-linux.tar.gz #NCBI官网下载源,同时下载md5校检码
md5sum -c ncbi-blast-2.14.0+-x64-linux.tar.gz.md5 #校检
tar -zxvf ncbi-blast-2.14.0+-x64-linux.tar.gz #解压缩
mv ncbi-blast-2.14.0+ blast #修改软件目录名字
export PATH=/home/blast/bin:$PATH #添加环境变量04 使用
进入/bin,里面有各种调用软件,具体如下
blastdb_aliastool blastdbcmd blast_formatter_vdb blastn_vdb blast_vdb_cmd cleanup-blastdb-volumes.py deltablast get_species_taxids.sh makeblastdb makeprofiledb rpsblast segmasker tblastn_vdb update_blastdb.pl
blastdbcheck blast_formatter blastn blastp blastx convert2blastmask dustmasker legacy_blast.pl makembindex psiblast rpstblastn tblastn tblastx windowmasker
最重要的3个程序:
1 makeblastdb
2 blastp
3 blastn
下面对这三个命令进行梳理4.1 检索库的建立
./makeblastdb -h
用法:
makeblastdb [-h] [-help] [-in 输入文件] [-input_type 类型]
-dbtype 分子类型 [-title 数据库标题] [-parse_seqids]
[-hash_index] [-mask_data 掩码数据文件] [-mask_id 掩码算法ID]
[-mask_desc 掩码算法描述] [-gi_mask]
[-gi_mask_name 基于GI的掩码名称] [-out 数据库名称]
[-blastdb_version 版本] [-max_file_sz 字节数]
[-metadata_output_prefix] [-logfile 文件名] [-taxid TaxID]
[-taxid_map TaxIDMapFile] [-oid_masks oid_masks] [-version]
*** 输入选项 #常用参数
-in <File_In>
输入文件/数据库名称
默认值 = `-'
-input_type <String, `asn1_bin', `asn1_txt', `blastdb`, `fasta'>
指定输入文件的数据类型
默认值 = `fasta'
*** 配置选项 #这部分通常可以忽略,也有说使用这些配置可以加快速度,那为什么不使用diamond呢?详细信息将在合集后续更新中提供,包括diamond的使用
-title <String>
BLAST数据库的标题
默认值 = 使用-in参数提供的输入文件名
-parse_seqids
如果设置,用于解析FASTA输入的seqid;对于所有其他输入类型,seqids会自动解析
-hash_index
创建序列哈希值索引。
*** 序列掩码选项 #可以忽略
-mask_data <String>
由NCBI掩码应用程序产生的掩码数据的输入文件列表,用逗号分隔(例如dustmasker, segmasker, windowmasker)
-mask_id <String>
用逗号分隔的字符串列表,唯一标识掩码算法
* 需要:mask_data
* 与:gi_mask不兼容
-mask_desc <String>
用逗号分隔的自由格式字符串列表,描述掩码算法的细节
* 需要:mask_id
-gi_mask
创建基于GI的掩码数据。
* 需要:parse_seqids
* 与:mask_id不兼容
-gi_mask_name <String>
掩码数据输出文件的列表,用逗号分隔。
* 需要:mask_data, gi_mask
*** 输出选项 ##重点参数
-out <String>
要创建的BLAST数据库的名称
默认值 = 使用-in参数提供的输入文件名。如果提供了多个文件/数据库作为输入,则此参数是必需的。
-blastdb_version <Integer, 4..5>
要创建的BLAST数据库的版本
默认值 = `5'
-max_file_sz <String>
BLAST数据库文件的最大文件大小
默认值 = `3GB'
-metadata_output_prefix <String>
数据库文件在元数据中的位置路径前缀
-logfile <File_Out>
程序日志应重定向到的文件
*** 分类选项 ##可以忽略
-taxid <Integer, >=0>
分配给所有序列的分类ID
* 与:taxid_map不兼容
-taxid_map <File_In>
将序列ID映射到分类ID的文本文件。
格式:<SequenceId> <TaxonomyId><换行>
* 需要:parse_seqids
* 与:taxid不兼容
-oid_masks <Integer>
0x01 排除模型
4.2 BLAST序列比对
4.2.1 氨基酸序列比对
./blastp -h
用法:
blastp [-h] [-help] [-import_search_strategy 文件名]
[-export_search_strategy 文件名] [-task 任务名称] [-db 数据库名称]
[-dbsize 字母数] [-gilist 文件名] [-seqidlist 文件名]
[-negative_gilist 文件名] [-negative_seqidlist 文件名]
[-taxids 税号] [-negative_taxids 税号] [-taxidlist 文件名]
[-negative_taxidlist 文件名] [-ipglist 文件名]
[-negative_ipglist 文件名] [-entrez_query entrez查询]
[-db_soft_mask 过滤算法] [-db_hard_mask 过滤算法]
[-subject 主题输入文件] [-subject_loc 范围] [-query 输入文件]
[-out 输出文件] [-evalue e值] [-word_size 整数值]
[-gapopen 开口罚分] [-gapextend 延伸罚分]
[-qcov_hsp_perc 浮点值] [-max_hsps 整数值]
[-xdrop_ungap 浮点值] [-xdrop_gap 浮点值]
[-xdrop_gap_final 浮点值] [-searchsp 整数值] [-seg SEG选项]
[-soft_masking 软掩码] [-matrix 矩阵名称]
[-threshold 浮点值] [-culling_limit 整数值]
[-best_hit_overhang 浮点值] [-best_hit_score_edge 浮点值]
[-subject_besthit] [-window_size 整数值] [-lcase_masking]
[-query_loc 范围] [-parse_deflines] [-outfmt 格式] [-show_gis]
[-num_descriptions 整数值] [-num_alignments 整数值]
[-line_length 行长度] [-html] [-sorthits 排序命中]
[-sorthsps 排序hps] [-max_target_seqs 序列数]
[-num_threads 整数值] [-mt_mode 整数值] [-ungapped] [-remote]
[-comp_based_stats 组成] [-use_sw_tback] [-version]
描述:
蛋白质-蛋白质BLAST 2.14.0+
可选参数:
-h
打印用法和描述;忽略所有其他参数
-help
打印用法、描述和参数;忽略所有其他参数
-version
打印版本号;忽略其他参数
*** 输入查询选项 #最重要的部分,这里是输入我们的查询序列
-query <File_In>
输入文件名
默认值 = `-'
-query_loc <String>
查询序列上的位置,使用1-based偏移量(格式:起始-终止)
*** 一般搜索选项 #这是搜索我们的上文件里的数据库,核心之二
-task <String, 允许的值: 'blastp' 'blastp-fast' 'blastp-short' >
要执行的任务
默认值 = `blastp'
-db <String>
BLAST数据库名称
* 与:subject, subject_loc不兼容
-out <File_Out, 文件名长度 < 256>
输出文件名
默认值 = `-'
-evalue <Real>
保存命中的期望值(E)阈值,默认为10
-word_size <Integer, >=2>
wordfinder算法的字大小
-gapopen <Integer>
开启间隙的成本
-gapextend <Integer>
延伸间隙的成本
-matrix <String>
计分矩阵名称(通常为BLOSUM62)
-threshold <Real, >=0>
最小单词分数,使得该单词被添加到BLAST查找表中
-comp_based_stats <String>
使用基于组成的统计:
D或d:默认(等同于2)
0或F或f:无基于组成的统计
1:如NAR 29:2994-3005, 2001中的基于组成的统计
2或T或t:如Bioinformatics 21:902-911, 2005中的基于组成的分数调整,基于序列属性
3:如Bioinformatics 21:902-911, 2005中的基于组成的分数调整,无条件
默认值 = `2'
*** BLAST-2-序列选项 #可忽略
-subject <File_In>
要搜索的主题序列
* 与:db, gilist, seqidlist, negative_gilist, negative_seqidlist, taxids, taxidlist, negative_taxids, negative_taxidlist, ipglist, negative_ipglist, db_soft_mask, db_hard_mask不兼容
-subject_loc <String>
主题序列上的位置,使用1-based偏移量(格式:起始-终止)
* 与:db, gilist, seqidlist, negative_gilist, negative_seqidlist, taxids, taxidlist, negative_taxids, negative_taxidlist, ipglist, negative_ipglist, db_soft_mask, db_hard_mask, remote不兼容
*** 格式化选项
-outfmt <String> #输出格式,核心之三,一般输出6
对齐视图选项:
0 = 成对,
1 = 查询锚定显示同一性,
2 = 查询锚定无同一性,
3 = 扁平查询锚定显示同一性,
4 = 扁平查询锚定无同一性,
5 = BLAST XML,
6 = 制表符分隔,
7 = 带注释行的制表符分隔,
8 = Seqalign (文本ASN.1),
9 = Seqalign (二进制ASN.1),
10 = 逗号分隔值,
11 = BLAST存档 (ASN.1),
12 = Seqalign (JSON),
13 = 多文件BLAST JSON,
14 = 多文件BLAST XML2,
15 = 单文件BLAST JSON,
16 = 单文件BLAST XML2,
18 = 有机体报告
选项6、7和10可以进一步配置以产生自定义格式,由空格分隔的格式说明符指定,或由delim关键字指定的令牌指定。
例如:"10 delim=@ qacc sacc score"。
delim关键字必须出现在数字输出格式规范之后。
支持的格式说明符包括:
qseqid 表示查询Seq-id
qgi 表示查询GI
qacc 表示查询准入
qaccver 表示查询准入版本
qlen 表示查询序列长度
sseqid 表示主题Seq-id
sallseqid 表示所有主题Seq-id(s),用';'分隔
sgi 表示主题GI
sallgi 表示所有主题GIs
sacc 表示主题准入
saccver 表示主题准入版本
sallacc 表示所有主题准入
slen 表示主题序列长度
qstart 表示查询中对齐的开始
qend 表示查询中对齐的结束
sstart 表示主题中对齐的开始
send 表示主题中对齐的结束
qseq 表示对齐的查询序列部分
sseq 表示对齐的主题序列部分
evalue 表示期望值
bitscore 表示比特分数
score 表示原始分数
length 表示对齐长度
pident 表示完全匹配的百分比
nident 表示完全匹配的数量
mismatch 表示不匹配的数量
positive 表示积极评分匹配的数量
gapopen 表示间隙开口的数量
gaps 表示总间隙的数量
ppos 表示积极评分匹配的百分比
frames 表示查询和主题框架,由'/'分隔
qframe 表示查询框架
sframe 表示主题框架
btop 表示Blast回溯操作 (BTOP)
staxid 表示主题分类ID
ssciname 表示主题科学名称
scomname 表示主题常见名称
sblastname 表示主题Blast名称
sskingdom 表示主题超级王国
staxids 表示唯一的主题分类ID(s),用';'分隔(按数字顺序)
sscinames 表示唯一的主题科学名称(s),用';'分隔
scomnames 表示唯一的主题常见名称(s),用';'分隔
sblastnames 表示唯一的主题Blast名称(s),用';'分隔(按字母顺序)
sskingdoms 表示唯一的主题超级王国(s),用';'分隔(按字母顺序)
stitle 表示主题标题
salltitles 表示所有主题标题(s),用'<>'分隔
sstrand 表示主题链
qcovs 表示查询覆盖率每个主题
qcovhsp 表示查询覆盖率每个HSP
qcovus 表示查询覆盖率每个独特主题(仅blastn)
如果未提供,默认值为:
'qaccver saccver pident length mismatch gapopen qstart qend sstart send evalue bitscore',等同于关键字'std'
默认值 = `0'
-show_gis
在deflines中显示NCBI GIs吗?
-num_descriptions <Integer, >=0>
显示一行描述的数据库序列数量
对于outfmt > 4不适用
默认值 = `500'
* 与:max_target_seqs不兼容
-num_alignments <Integer, >=0>
显示对齐的数据库序列数量
默认值 = `250'
* 与:max_target_seqs不兼容
-line_length <Integer, >=1>
格式化对齐的行长度
对于outfmt > 4不适用
默认值 = `60'
-html
产生HTML输出?
-sorthits <Integer, (>=0 and =<4)>
命中的排序选项:
对齐视图选项:
0 = 按evalue排序,
1 = 按比特分数排序,
2 = 按总分排序,
3 = 按百分比同一性排序,
4 = 按查询覆盖率排序
对于outfmt > 4不适用
-sorthsps <Integer, (>=0 and =<4)>
hps的排序选项:
0 = 按hsp evalue排序,
1 = 按hsp分数排序,
2 = 按hsp查询开始排序,
3 = 按hsp百分比同一性排序,
4 = 按hsp主题开始排序
对于outfmt != 0不适用
*** 查询过滤选项 #可忽略
-seg <String>
使用SEG过滤查询序列(格式:'yes', 'window locut hicut', 或 'no'禁用)
默认值 = `no'
-soft_masking <Boolean>
将过滤位置应用为软掩码
默认值 = `false'
-lcase_masking
在查询和主题序列中使用小写过滤?
*** 限制搜索或结果
-gilist <String>
将数据库搜索限制为GI列表
* 与:seqidlist, taxids, taxidlist, negative_gilist, negative_seqidlist, negative_taxids, negative_taxidlist, remote, subject, subject_loc不兼容
-seqidlist <String>
将数据库搜索限制为SeqID列表
* 与:gilist, taxids, taxidlist, negative_gilist, negative_seqidlist, negative_taxids, negative_taxidlist, remote, subject, subject_loc不兼容
-negative_gilist <String>
将数据库搜索限制为除指定GI之外的所有内容
* 与:gilist, seqidlist, taxids, taxidlist, negative_seqidlist, negative_taxids, negative_taxidlist, remote, subject, subject_loc不兼容
-negative_seqidlist <String>
将数据库搜索限制为除指定SeqID之外的所有内容
* 与:gilist, seqidlist, taxids, taxidlist, negative_gilist, negative_taxids, negative_taxidlist, remote, subject, subject_loc不兼容
-taxids <String>
将数据库搜索限制为仅包括指定的分类ID(多个ID由','分隔)
* 与:gilist, seqidlist, taxidlist, negative_gilist, negative_seqidlist, negative_taxids, negative_taxidlist, remote, subject, subject_loc不兼容
-negative_taxids <String>
将数据库搜索限制为除指定分类ID之外的所有内容(多个ID由','分隔)
* 与:gilist, seqidlist, taxids, taxidlist, negative_gilist, negative_seqidlist, negative_taxidlist, remote, subject, subject_loc不兼容
-taxidlist <String>
将数据库搜索限制为仅包括指定的分类ID
* 与:gilist, seqidlist, taxids, negative_gilist, negative_seqidlist, negative_taxids, negative_taxidlist, remote, subject, subject_loc不兼容
-negative_taxidlist <String>
将数据库搜索限制为除指定分类ID之外的所有内容
* 与:gilist, seqidlist, taxids, taxidlist, negative_gilist, negative_seqidlist, negative_taxids, remote, subject, subject_loc不兼容
-ipglist <String>
将数据库搜索限制为IPG列表
* 与:subject, subject_loc不兼容
-negative_ipglist <String>
将数据库搜索限制为除指定IPG之外的所有内容
* 与:subject, subject_loc不兼容
-entrez_query <String>
使用给定的Entrez查询限制搜索
* 需要:remote
-db_soft_mask <String>
将过滤算法ID应用于BLAST数据库作为软掩码
* 与:db_hard_mask, subject, subject_loc不兼容
-db_hard_mask <String>
将过滤算法ID应用于BLAST数据库作为硬掩码
* 与:db_soft_mask, subject, subject_loc不兼容
-qcov_hsp_perc <Real, 0..100>
每个hsp的查询覆盖率百分比
-max_hsps <Integer, >=1>
为每个查询保存每个主题序列的最大HSP数量
-culling_limit <Integer, >=0>
如果一个命中的查询范围被至少这么多更高分数的命中包围,删除该命中
* 与:best_hit_overhang, best_hit_score_edge不兼容
-best_hit_overhang <Real, (>0 and <0.5)>
最佳命中算法的悬垂值(推荐值:0.1)
* 与:culling_limit不兼容
-best_hit_score_edge <Real, (>0 and <0.5)>
最佳命中算法的分数边缘值(推荐值:0.1)
* 与:culling_limit不兼容
-subject_besthit
开启每个主题序列的最佳命中
-max_target_seqs <Integer, >=1>
保留的最大对齐序列数量
(推荐值为5或更多)
默认值 = `500'
* 与:num_descriptions, num_alignments不兼容
*** 统计选项 #可忽略
-dbsize <Int8>
数据库的有效长度
-searchsp <Int8, >=0>
搜索空间的有效长度
*** 搜索策略选项
-import_search_strategy <File_In>
使用的搜索策略
* 与:export_search_strategy不兼容
-export_search_strategy <File_Out>
记录所使用搜索策略的文件名
* 与:import_search_strategy不兼容
*** 扩展选项
-xdrop_ungap <Real>
无间隙扩展的X-dropoff值(以比特为单位)
-xdrop_gap <Real>
初步有间隙扩展的X-dropoff值(以比特为单位)
-xdrop_gap_final <Real>
最终有间隙对齐的X-dropoff值(以比特为单位)
-window_size <Integer, >=0>
多重命中窗口大小,使用0指定1-hit算法
-ungapped
仅执行无间隙对齐?
*** 其他选项 #可忽略
-parse_deflines
是否解析查询和主题的defline(s)?
-num_threads <Integer, >=1>
在BLAST搜索中使用的线程(CPU)数量
默认值 = `1'
* 与:remote不兼容
-mt_mode <Integer, (>=0 and =<1)>
在BLAST搜索中使用的多线程模式:
0(自动)按数据库分割
1按查询分割
默认值 = `0'
* 需要:num_threads
-remote
远程执行搜索?
* 与:gilist, seqidlist, taxids, taxidlist, negative_gilist, negative_seqidlist, negative_taxids, negative_taxidlist, subject_loc, num_threads不兼容
-use_sw_tback
计算局部最优的Smith-Waterman对齐?4.2.2 核酸序列比对

./blastn -h
用法:
blastn [-h] [-help] [-import_search_strategy 文件名]
[-export_search_strategy 文件名] [-task 任务名称] [-db 数据库名称]
[-dbsize 字母数] [-gilist 文件名] [-seqidlist 文件名]
[-negative_gilist 文件名] [-negative_seqidlist 文件名]
[-taxids 税号] [-negative_taxids 税号] [-taxidlist 文件名]
[-negative_taxidlist 文件名] [-entrez_query entrez查询]
[-db_soft_mask 过滤算法] [-db_hard_mask 过滤算法]
[-subject 主题输入文件] [-subject_loc 范围] [-query 输入文件]
[-out 输出文件] [-evalue e值] [-word_size 整数值]
[-gapopen 开口罚分] [-gapextend 延伸罚分]
[-perc_identity 浮点值] [-qcov_hsp_perc 浮点值]
[-max_hsps 整数值] [-xdrop_ungap 浮点值] [-xdrop_gap 浮点值]
[-xdrop_gap_final 浮点值] [-searchsp 整数值] [-penalty 罚分]
[-reward 奖励] [-no_greedy] [-min_raw_gapped_score 整数值]
[-template_type 类型] [-template_length 整数值] [-dust DUST选项]
[-filtering_db 过滤数据库]
[-window_masker_taxid window_masker_taxid]
[-window_masker_db window_masker_db] [-soft_masking 软掩码]
[-ungapped] [-culling_limit 整数值] [-best_hit_overhang 浮点值]
[-best_hit_score_edge 浮点值] [-subject_besthit]
[-window_size 整数值] [-off_diagonal_range 整数值]
[-use_index 布尔值] [-index_name 字符串] [-lcase_masking]
[-query_loc 范围] [-strand 链] [-parse_deflines] [-outfmt 格式]
[-show_gis] [-num_descriptions 整数值] [-num_alignments 整数值]
[-line_length 行长度] [-html] [-sorthits 排序命中]
[-sorthsps 排序HSPs] [-max_target_seqs 序列数]
[-num_threads 整数值] [-mt_mode 整数值] [-remote] [-version]
描述:
核酸-核酸BLAST 2.14.0+
可选参数:
-h
打印用法和描述;忽略所有其他参数
-help
打印用法、描述和参数;忽略所有其他参数
-version
打印版本号;忽略其他参数
*** 输入查询选项
-query <File_In> #核心参数之一,查询的内容
输入文件名
默认值 = `-'
-query_loc <String>
查询序列上的位置,使用1-based偏移量(格式:起始-终止)
-strand <String, `both', `minus', `plus'>
针对数据库/主题搜索的查询链
默认值 = `both'
*** 一般搜索选项 #核心参数之二,上文建立的数据库
-task <String, 允许的值: 'blastn' 'blastn-short' 'dc-megablast' 'megablast' 'rmblastn'>
要执行的任务
默认值 = `megablast'
-db <String>
BLAST数据库名称
* 与:subject, subject_loc不兼容
-out <File_Out, 文件名长度 < 256>
输出文件名
默认值 = `-'
-evalue <Real>
保存命中的期望值(E)阈值,默认为10(对于blastn-short为1000)
-word_size <Integer, >=4>
wordfinder算法的字大小(最佳完美匹配的长度)
-gapopen <Integer>
开启间隙的成本
-gapextend <Integer>
延伸间隙的成本
-penalty <Integer, <=0>
核酸不匹配的罚分
-reward <Integer, >=0>
核酸匹配的奖励
-use_index <Boolean>
使用MegaBLAST数据库索引
默认值 = `false'
-index_name <String>
MegaBLAST数据库索引名称(已弃用;仅用于旧式索引)
*** BLAST-2-序列选项 #可忽略
-subject <File_In>
要搜索的主题序列
* 与:db, gilist, seqidlist, negative_gilist, negative_seqidlist, taxids, taxidlist, negative_taxids, negative_taxidlist, db_soft_mask, db_hard_mask不兼容
-subject_loc <String>
主题序列上的位置,使用1-based偏移量(格式:起始-终止)
* 与:db, gilist, seqidlist, negative_gilist, negative_seqidlist, taxids, taxidlist, negative_taxids, negative_taxidlist, db_soft_mask, db_hard_mask, remote不兼容
*** 格式化选项 ##核心参数之三,一般选择outfmt-6格式
-outfmt <String>
对齐视图选项:
0 = 成对,
1 = 查询锚定显示同一性,
2 = 查询锚定无同一性,
3 = 扁平查询锚定显示同一性,
4 = 扁平查询锚定无同一性,
5 = BLAST XML,
6 = 制表符分隔,
7 = 带注释行的制表符分隔,
8 = Seqalign (文本ASN.1),
9 = Seqalign (二进制ASN.1),
10 = 逗号分隔值,
11 = BLAST存档 (ASN.1),
12 = Seqalign (JSON),
13 = 多文件BLAST JSON,
14 = 多文件BLAST XML2,
15 = 单文件BLAST JSON,
16 = 单文件BLAST XML2,
17 = 序列对齐/映射 (SAM),
18 = 有机体报告
选项6、7、10和17可以进一步配置以产生自定义格式,由空格分隔的格式说明符指定,或在选项6、7和10的情况下,由delim关键字指定的令牌指定。例如:"17 delim=@ qacc sacc score"。
delim关键字必须出现在数字输出格式规范之后。
支持的格式说明符包括:
qseqid 表示查询Seq-id
qgi 表示查询GI
qacc 表示查询准入
qaccver 表示查询准入版本
qlen 表示查询序列长度
sseqid 表示主题Seq-id
sallseqid 表示所有主题Seq-id(s),用';'分隔
sgi 表示主题GI
sallgi 表示所有主题GIs
sacc 表示主题准入
saccver 表示主题准入版本
sallacc 表示所有主题准入
slen 表示主题序列长度
qstart 表示查询中对齐的开始
qend 表示查询中对齐的结束
sstart 表示主题中对齐的开始
send 表示主题中对齐的结束
qseq 表示对齐的查询序列部分
sseq 表示对齐的主题序列部分
evalue 表示期望值
bitscore 表示比特分数
score 表示原始分数
length 表示对齐长度
pident 表示完全匹配的百分比
nident 表示完全匹配的数量
mismatch 表示不匹配的数量
positive 表示积极评分匹配的数量
gapopen 表示间隙开口的数量
gaps 表示总间隙的数量
ppos 表示积极评分匹配的百分比
frames 表示查询和主题框架,由'/'分隔
qframe 表示查询框架
sframe 表示主题框架
btop 表示Blast回溯操作 (BTOP)
staxid 表示主题分类ID
ssciname 表示主题科学名称
scomname 表示主题常见名称
sblastname 表示主题Blast名称
sskingdom 表示主题超级王国
staxids 表示唯一的主题分类ID(s),用';'分隔(按数字顺序)
sscinames 表示唯一的主题科学名称(s),用';'分隔
scomnames 表示唯一的主题常见名称(s),用';'分隔
sblastnames 表示唯一的主题Blast名称(s),用';'分隔(按字母顺序)
sskingdoms 表示唯一的主题超级王国(s),用';'分隔(按字母顺序)
stitle 表示主题标题
salltitles 表示所有主题标题(s),用'<>'分隔
sstrand 表示主题链
qcovs 表示查询覆盖率每个主题
qcovhsp 表示查询覆盖率每个HSP
qcovus 表示查询覆盖率每个独特主题(仅blastn)
如果未提供,默认值为:
'qaccver saccver pident length mismatch gapopen qstart qend sstart send evalue bitscore',等同于关键字'std'
-show_gis
在deflines中显示NCBI GIs吗?
-num_descriptions <Integer, >=0>
显示一行描述的数据库序列数量
对于outfmt > 4不适用
默认值 = `500'
* 与:max_target_seqs不兼容
-num_alignments <Integer, >=0>
显示对齐的数据库序列数量
默认值 = `250'
* 与:max_target_seqs不兼容
-line_length <Integer, >=1>
格式化对齐的行长度
对于outfmt > 4不适用
默认值 = `60'
-html
产生HTML输出?
-sorthits <Integer, (>=0 and =<4)>
命中的排序选项:
对齐视图选项:
0 = 按evalue排序,
1 = 按比特分数排序,
2 = 按总分排序,
3 = 按百分比同一性排序,
4 = 按查询覆盖率排序
对于outfmt > 4不适用
-sorthsps <Integer, (>=0 and =<4)>
HSPs的排序选项:
0 = 按hsp evalue排序,
1 = 按hsp分数排序,
2 = 按hsp查询开始排序,
3 = 按hsp百分比同一性排序,
4 = 按hsp主题开始排序
对于outfmt != 0不适用
*** 查询过滤选项 #可忽略
-dust <String>
使用DUST过滤查询序列(格式:'yes', 'level window linker', 或 'no'禁用)默认值为'20 64 1'(对于blastn-short为'no')
-filtering_db <String>
包含过滤元素(例如:重复序列)的BLAST数据库
-window_masker_taxid <Integer>
使用Taxonomic ID启用WindowMasker过滤
-window_masker_db <String>
使用此重复序列数据库启用WindowMasker过滤。
-soft_masking <Boolean>
将过滤位置应用为软掩码
默认值 = `true'
-lcase_masking
在查询和主题序列中使用小写过滤?
*** 限制搜索或结果
-gilist <String>
将数据库搜索限制为GI列表
* 与:seqidlist, taxids, taxidlist, negative_gilist, negative_seqidlist, negative_taxids, negative_taxidlist, remote, subject, subject_loc不兼容
-seqidlist <String>
将数据库搜索限制为SeqID列表
* 与:gilist, taxids, taxidlist, negative_gilist, negative_seqidlist, negative_taxids, negative_taxidlist, remote, subject, subject_loc不兼容
-negative_gilist <String>
将数据库搜索限制为除指定GI之外的所有内容
* 与:gilist, seqidlist, taxids, taxidlist, negative_seqidlist, negative_taxids, negative_taxidlist, remote, subject, subject_loc不兼容
-negative_seqidlist <String>
将数据库搜索限制为除指定SeqID之外的所有内容
* 与:gilist, seqidlist, taxids, taxidlist, negative_gilist, negative_taxids, negative_taxidlist, remote, subject, subject_loc不兼容
-taxids <String>
将数据库搜索限制为仅包括指定的分类ID(多个ID由','分隔)
* 与:gilist, seqidlist, taxidlist, negative_gilist, negative_seqidlist, negative_taxids, negative_taxidlist, remote, subject, subject_loc不兼容
-negative_taxids <String>
将数据库搜索限制为除指定分类ID之外的所有内容(多个ID由','分隔)
* 与:gilist, seqidlist, taxids, taxidlist, negative_gilist, negative_seqidlist, negative_taxidlist, remote, subject, subject_loc不兼容
-taxidlist <String>
将数据库搜索限制为仅包括指定的分类ID
* 与:gilist, seqidlist, taxids, negative_gilist, negative_seqidlist, negative_taxids, negative_taxidlist, remote, subject, subject_loc不兼容
-negative_taxidlist <String>
将数据库搜索限制为除指定分类ID之外的所有内容
* 与:gilist, seqidlist, taxids, taxidlist, negative_gilist, negative_seqidlist, negative_taxids, remote, subject, subject_loc不兼容
-entrez_query <String>
使用给定的Entrez查询限制搜索
* 需要:remote
-db_soft_mask <String>
将过滤算法ID应用于BLAST数据库作为软掩码
* 与:db_hard_mask, subject, subject_loc不兼容
-db_hard_mask <String>
将过滤算法ID应用于BLAST数据库作为硬掩码
* 与:db_soft_mask, subject, subject_loc不兼容
-perc_identity <Real, 0..100>
同一性百分比
-qcov_hsp_perc <Real, 0..100>
每个hsp的查询覆盖率百分比
-max_hsps <Integer, >=1>
为每个查询保存每个主题序列的最大HSP数量
-culling_limit <Integer, >=0>
如果一个命中的查询范围被至少这么多更高分数的命中包围,删除该命中
* 与:best_hit_overhang, best_hit_score_edge不兼容
-best_hit_overhang <Real, (>0 and <0.5)>
最佳命中算法的悬垂值(推荐值:0.1)
* 与:culling_limit不兼容
-best_hit_score_edge <Real, (>0 and <0.5)>
最佳命中算法的分数边缘值(推荐值:0.1)
* 与:culling_limit不兼容
-subject_besthit
开启每个主题序列的最佳命中
-max_target_seqs <Integer, >=1>
保留的最大对齐序列数量
(推荐值为5或更多)
默认值 = `500'
* 与:num_descriptions, num_alignments不兼容
*** 不连续MegaBLAST选项
-template_type <String, `coding', `coding_and_optimal', `optimal'>
不连续MegaBLAST模板类型
* 需要:template_length
-template_length <Integer, 允许的值: '16' '18' '21'>
不连续MegaBLAST模板长度
* 需要:template_type
*** 统计选项 #可忽略
-dbsize <Int8>
数据库的有效长度
-searchsp <Int8, >=0>
搜索空间的有效长度
*** 搜索策略选项
-import_search_strategy <File_In>
使用的搜索策略
* 与:export_search_strategy不兼容
-export_search_strategy <File_Out>
记录所使用搜索策略的文件名
* 与:import_search_strategy不兼容
*** 扩展选项 #可忽略
-xdrop_ungap <Real>
无间隙扩展的X-dropoff值(以比特为单位)
-xdrop_gap <Real>
初步有间隙扩展的X-dropoff值(以比特为单位)
-xdrop_gap_final <Real>
最终有间隙对齐的X-dropoff值(以比特为单位)
-no_greedy
使用非贪婪的动态规划扩展
-min_raw_gapped_score <Integer>
在初步有间隙和回溯阶段保留对齐的最小原始分数
-ungapped
仅执行无间隙对齐?
-window_size <Integer, >=0>
多重命中窗口大小,使用0指定1-hit算法
-off_diagonal_range <Integer, >=0>
搜索第二次命中的偏对角线数量,使用0关闭
默认值 = `0'
*** 其他选项 #可忽略
-parse_deflines
是否解析查询和主题的defline(s)?
-num_threads <Integer, >=1>
在BLAST搜索中使用的线程(CPU)数量
默认值 = `1'
* 与:remote不兼容
-mt_mode <Integer, (>=0 and =<1)>
在BLAST搜索中使用的多线程模式:
0(自动)按数据库分割
1按查询分割
默认值 = `0'
* 需要:num_threads
-remote
远程执行搜索?
* 与:gilist, seqidlist, taxids, taxidlist, negative_gilist, negative_seqidlist, negative_taxids, negative_taxidlist, subject_loc, num_threads不兼容05 常用命令行
5.1 blastn and blastp
(1) 建立数据库(核酸库or蛋白库)
makeblastdb -in db.fasta -parse_seqids -hash_index -dbtype prot -out pep.fasta.db
#-in参数后面接将要格式化的数据库;
#-parse_seqids, -hash_index两个参数一般都带上,主要是为blastdbcmd取子序列时使用;
#-dbtype 后接所格式化的序列的类型,核酸用 nucl,蛋白质用prot;
#-out 后接数据库名,自己起一个有意义的名字,后面比对搜索时要用到的-db的参数。
(2) BLAST序列比对
blastn -query cds.fasta -out cds.blast.outfmt6 -db cds.fasta.db -outfmt 6 -evalue 1e-5 -num_threads 10
blastp -query pep.fasta -out pep.blast.outfmt6 -db pep.fasta.db -outfmt 6 -evalue 1e-5 -num_threads 10
#-query: 输入文件路径及文件名;
#-out:输出文件路径及文件名;
#-db:数据库路径及数据库名,注意区分核酸库和蛋白库;
#-evalue:设置输出结果的e-value值,一般1e-5,这其实也是默认值;
#-num_threads:线程数,服务器用10线程适中。
(3) 结果解读
outfmt 6输出12列对应的含义:
Query id:查询序列ID标识;
Subject id:比对上的目标序列ID标识;
% identity:序列比对的一致性百分比;
alignment length:符合比对的比对区域的长度;
mismatches:比对区域的错配数;
gap openings:比对区域的gap数目;
q.start:比对区域在查询序列(Query id)上的起始位点;
q.end:比对区域在查询序列(Query id)上的终止位点;
s.start:比对区域在目标序列(Subject id)上的起始位点;
s.end:比对区域在目标序列(Subject id)上的终止位点;
e-value:比对结果的期望值,解释是大概多少次随即比对才能出现一次这个score,Evalue越小,表明这种情况,从概率上越不可能发生,但是现在发生了,所以这个比对具有很重要的意义;
bit score:比对结果的bit score值。bit score值表示对齐的好坏;分数越高,对齐效果越好。一般来说,这个分数是根据一个公式计算出来的,这个公式考虑了相似或相同残基的排列,以及为了排列序列而引入的任何gaps。
less pep.blast.outfmt6
cat pep.blast.outfmt6
对于结果即第11列越小,第十二列越大越好!5.2 blastn、blastp、blastx、tblastn和tblastx的区别与用法
BLAST(Basic Local Alignment Search Tool)是一种用于比较基因和蛋白质序列的工具,它有多种变体,分别适用于不同的序列比较任务。以下是BLAST各变体的区别与用法:
5.2.1 BLASTN
用途:用于比较核酸序列(如DNA或RNA)。
数据库:核酸序列数据库。
查询序列:核酸序列。
典型用法:
- 用于在基因组数据库中查找与给定DNA序列相似的序列。
- 用于基因定位、基因注释和识别保守区域。
5.2.2 BLASTP
用途:用于比较蛋白质序列。
数据库:蛋白质序列数据库。
查询序列:蛋白质序列。
典型用法:
- 用于在蛋白质数据库中查找与给定蛋白质序列相似的序列。
- 用于预测蛋白质功能、识别蛋白质家族和结构域。
5.2.3 BLASTX
用途:用于将核酸序列翻译为蛋白质序列后进行比较。
数据库:蛋白质序列数据库。
查询序列:核酸序列(翻译为蛋白质序列)。
典型用法:
- 用于预测基因编码区域,特别是在全基因组或转录组数据中。
- 用于将未知的DNA序列与已知的蛋白质序列进行比较,推断编码蛋白质的功能。
5.2.4 TBLASTN
用途:用于将蛋白质序列与翻译的核酸序列进行比较。
数据库:核酸序列数据库(翻译为蛋白质序列)。
查询序列:蛋白质序列。
典型用法:
- 用于在基因组数据库中查找与给定蛋白质序列相似的基因。
- 用于识别基因组中可能编码特定蛋白质的区域。
5.2.5 TBLASTX
用途:用于将翻译后的核酸序列与翻译后的核酸序列进行比较。
数据库:核酸序列数据库(翻译为蛋白质序列)。
查询序列:核酸序列(翻译为蛋白质序列)。
典型用法:
- 用于在基因组数据库中查找与给定DNA序列相似的序列,且这些序列被翻译为蛋白质。
- 用于跨物种或不同基因组之间的比较基因组学研究。
用法示例
1. BLASTN:
blastn -query my_dna_sequence.fasta -db nt -out results_blastn.txt
2. BLASTP:
blastp -query my_protein_sequence.fasta -db nr -out results_blastp.txt
3. BLASTX:
blastx -query my_dna_sequence.fasta -db nr -out results_blastx.txt
4. TBLASTN:
tblastn -query my_protein_sequence.fasta -db nt -out results_tblastn.txt
5. TBLASTX:
tblastx -query my_dna_sequence.fasta -db nt -out results_tblastx.txt
通过选择合适的BLAST工具,可以有效地进行序列比对和分析,满足不同的生物信息学研究需求。
他确实比diamond的功能更多,经典的还是最全的!
diamond安装与使用-diamond-2.1.8(bioinfomatics tools-010)
BLAST一般在国内生物信息学专业教材中数据库和序列比对相关章节,并且众多生物化学和分子生物学教材中基因组与比较基因组学相关内容也已单独成章。通过测序获得基因、甚至基因组序列,也已成为分子生物学实验室的常规实验方法。
生信的入门一定绕不开blast,祝君且前行!
06 参考文献
McGinnis, S., & Madden, T. L. (2004). BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic acids research, 32(Web Server issue), W20–W25. https://doi.org/10.1093/nar/gkh435
Al-Fatlawi, A., Menzel, M. & Schroeder, M. Is Protein BLAST a thing of the past?. Nat Commun 14, 8195 (2023). https://doi.org/10.1038/s41467-023-44082-5
Cao, ZJ., Wei, L., Lu, S. et al. Searching large-scale scRNA-seq databases via unbiased cell embedding with Cell BLAST. Nat Commun 11, 3458 (2020). https://doi.org/10.1038/s41467-020-17281-7
罗静初.双序列比对基础和应用实例[J].生物信息学,2023,21(01):1-19.















 1069
1069

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









