










 本文结合长读测序和Hi - C支架,为毛白杨提供全新组装。研究确定其基因组大小、结构,推断祖先为母本杨和父本白杨变种毛白杨,发现基因组存在广泛结构变异,这些成果有助于阐明杨树物种形成机制,扩展对杨树基因组生物学的理解。
本文结合长读测序和Hi - C支架,为毛白杨提供全新组装。研究确定其基因组大小、结构,推断祖先为母本杨和父本白杨变种毛白杨,发现基因组存在广泛结构变异,这些成果有助于阐明杨树物种形成机制,扩展对杨树基因组生物学的理解。
High quality haplotype-resolved genome assemblies of Populus tomentosa Carr., a stabilized interspecific hybrid species widespread in Asia
高质量二倍体解析的毛白杨(Populus tomentosa Carr.)基因组组装,这是一种在亚洲广泛分布的稳定杂交种。

摘要
杨树具有广泛的生态地理分布,覆盖北半球,而且种间杂交很常见。毛白杨(Populus tomentosa Carr.)在亚洲东部地区广泛分布和栽培,它在林业、农业、保护和城市园艺中扮演多重重要角色。虽然已有几种杨树的参考基因组,但我们的目标是生成一个非常高质量的毛白杨染色体水平的全新基因组组装,这将作为整个属杂交物种形成的进化和生态研究的参考。在此,我们结合了长读测序和Hi-C支架,呈现了一个高质量的二倍体解析基因组组装。该基因组大小为740.2 Mb,contig N50大小为5.47 Mb,scaffold N50大小为46.68 Mb,由38条染色体组成,符合已知的二倍体染色体数(2n = 2x = 38)。共鉴定出59,124个蛋白编码基因。系统发生基因组分析显示,毛白杨由两个不同的亚基因组组成,我们证明这很可能是由母本杨(Populus adenopoda)和父本白杨变种毛白杨(Populus alba var. pyramidalis)杂交形成的,起源时间约为393万年前。尽管高度共线,但两个亚基因组之间发现了显著的结构变异。我们的研究为生态遗传学和森林生物技术提供了宝贵的资源。
1 引言
基因组学革命促使了在广泛的模型生物和非模型生物中对整个基因组的测序和组装的前所未有的增长(Ellegren, 2014)。虽然这推动了对适应性特征的遗传基础研究的大型基因组多样性面板的发展,但在一个物种内或一组近缘种内依赖单一的、组装良好的参考基因组对遗传和进化推断带来了重大限制(Sherman & Salzberg, 2020)。当处理大型、结构多样、杂交或杂合基因组时,这一挑战尤为严峻,因为使用不同的参考基因组映射短读序列可能会导致低覆盖和变异检测的偏差。这种复杂性挑战了植物基因组的组装,对于植物基因组来说,理想的是使用同质纯合系(Daccord et al., 2017; Wu et al., 2018)。然而,许多植物包括杨树本质上是异交的,因此杂合基因区域普遍存在,是表型变异的主要贡献者(Schnable & Springer, 2013)。此外,直接测序自然杂合系或人工驯化的杂交系可以深入了解它们的遗传复杂性和进化历史(Minio et al., 2019)。
杨属(包括白杨、杨树和白桦等)已成为树木生态基因组学和生物技术的领先模型,包括为杨树(Populus trichocarpa)——第一棵进行全基因组测序的树木——开发参考基因组组装(Tuskan et al., 2006)。近年来,也已公布了P. euphratica、P. tremula 及 tremuloides、P. alba var. pyramidalis 和 P. alba 的整个基因组(Lin et al., 2018; Liu et al., 2019; Ma et al., 2019; 2013a; 2013b)。然而,高遗传杂合性和第三代测序技术的有限应用限制了许多这些基因组组装的质量,这些组装通常仍然高度破碎,分成数千个支架(Ambardar et al., 2016)。
多个高度连续、组装良好的杨树参考基因组的可用性将大大促进准确推断同源性、重组和染色体起源(Lin et al., 2018)。多样化的、组装良好的参考基因组也将为这个经济重要属的功能基因组学、遗传工程和分子育种提供一个基本工具(Zhang et al., 2019)。它还将改进杨树泛基因组的系统发生基因组学分析(Pinosio et al., 2016; Zhang et al., 2019),无需依赖仅基于P. trichocarpa参考基因组的参考引导映射和变异检测。最近在整个基因组测序方法上的进步,包括染色体构象捕获(Hi-C;van Berkum et al., 2010)和长读测序,提供了一种超越破碎草图基因组和生成几乎全面的全新组装的方法(El-Metwally et al., 2014)。
毛白杨(Populus tomentosa),也称为中国白杨,原产并广泛分布于中国大部分地区(An et al., 2011)。此外,它也是中国大规模人工种植园中首批种植的树种。其特点包括快速生长、粗直的树干、高环境适应性和长寿命(通常100-200年,有时超过500年)。这些特性使得毛白杨从经济、生态和进化的角度都非常有价值,其应用包括木材、纸浆和纸张、单板、胶合板、生物修复、防风、固碳和防止水土流失。像其他白杨一样,毛白杨已成为树木遗传研究的重要模型(An et al., 2011),但目前尚无基因组序列可用,毛白杨的起源、进化和遗传结构尚不清楚。有人提出毛白杨是杨属中的一个独特物种(Dickmann & Isebrands, 2001)。然而,毛白杨的起源一直存在争议。尽管曾提出毛白杨包含两种具有不同母本的遗传类型(Wang et al., 2019),但关于杂交起源的建议是基于有限的分子标记和不完整的原产地材料集。因此,其祖先和基因组结构仍然不清楚。我们的研究通过提供对基因组结构和结构组成的更深入了解,增加了对该物种的了解,这些都是在推断的种间杂交后获得的。
在这里,我们通过结合应用PacBio、Illumina和Hi-C测序技术,为毛白杨(克隆GM15)提供全新组装。我们在此提供了所有染色体的两个高质量二倍体解析组装,其系统发生关系证明了这个物种的杂交起源。通过结合本研究中叶绿体和线粒体基因组的系统发生分析,我们推断毛白杨的祖先是P. adenopoda(母本)和P. alba var. pyramidalis(父本)。此外,我们还发现了基因组中广泛的结构变异。这些发现有助于阐明杨树物种形成的机制,并扩展了我们对杨树基因组生物学的理解。与此同时,毛白杨作为一种具有许多优良特性的稳定种间杂交物种,在亚洲广泛分布,揭示了亲本起源和高质量二倍体解析基因组,将引起更广泛社区的兴趣。
2 材料与方法
2.1 体外再生和验证
我们从优良的雄性毛白杨克隆(LM50)中采集带有花芽的枝条,并在温室中进行水培。随后,我们进行了花药诱导再生,并测量了再生花药植株的倍性(参考先前研究:[Li et al., 2013])。使用Cell Laboratory Quanta SC(Beckman Coulter)检测了细胞核DNA含量(C值)。在根尖细胞上进行染色体计数,并使用FSX-100显微镜相机系统(Olympus)拍摄了显微照片。同样处理的毛白杨(克隆LM50)叶子被用作二倍体对照。此外,使用位于19条染色体上的19个等位基因特异性引物对进行了花药植株的基因型鉴定(表S1)。
2.2 基因组DNA文库构建和测序
选择通过体外花药培养和再生系统生成的植株GM15进行基因组测序。使用Qiagen DNeasy Plant Mini Kit提取基因组DNA。通过琼脂糖凝胶电泳评估DNA质量,并使用NanoDrop分光光度计(Thermo Fisher Scientific)确定其数量。根据制造商的协议(Illumina Inc.)构建无PCR短插入基因组DNA文库(300-500 bp),用于Illumina HiSeq X Ten测序仪上的配对末端测序。对于SMRT测序,根据制造商的协议(Pacific Biosciences)构建20-kb基因组DNA文库。最终,50 μg高质量基因组DNA在PacBio Sequel平台(Pacific Biosciences)上进行测序,生成了11个SMRT细胞。为了比较GM15和LM50之间的kmer基因组特征,LM50也使用相同的程序在Illumina平台上进行了测序,包括DNA提取、文库构建和测序。
2.3 RNA-seq文库构建和测序
使用Qiagen RNeasy Plant Mini Kit(Qiagen)提取植株GM15的根、茎和叶的总RNA。通过琼脂糖凝胶电泳评估RNA质量,并使用NanoDrop分光光度计(Thermo Fisher Scientific)确定其数量。为了辅助基因注释,使用从提取的组织中得到的2 μg总RNA构建RNA-seq文库,并根据制造商的协议(New England Biolabs)在Illumina HiSeq X Ten平台上进行测序。
2.4 基因组大小估计和组装
使用基因组特性估计(GCE)程序v1.0(Liu et al., 2013),通过基于PCR-free Illumina短读的17-mer分析估计GM15和LM50的基因组大小。基于过滤后的PacBio读取(过滤掉小于1000 bp的读取后),使用overlap-layout-consensus方法在CANU v1.5(Koren et al., 2017)中进行了去 novo组装。随后,使用Arrow(https://github.com/PacificBiosciences/GenomicConsensus)对初步草图组装进行了打磨,以提高准确性。
2.5 Hi-C文库测序和染色体锚定
使用标准程序(NEBNext Ultra II DNA Library Prep Kit for Illumina)准备Hi-C文库,并在Illumina HiSeq X Ten平台上进行测序。Hi-C读数首先使用Juicer v1.5.6(Durand, Shamim等,2016)映射到上述草图基因组。为了考虑杨树中的高复制水平,只有映射质量>40的对齐读数被用来使用3D-DNA(v170123)流水线进行Hi-C关联染色体组装,参数为“-m haploid -t 5000 -s 2”(Dudchenko等,2017)。使用Juicebox v1.6(Durand, Robinson等,2016)进行可视化,得到包含38条染色体的染色体规模草图组装。随后,使用PacBio读数对组装进行了三次Arrow打磨,并使用Illumina短读与Pilon v1.22进行了五次打磨。最终,通过使用bowtie2 v2.3.4、BLASR v5.1(Chaisson & Tesler, 2012)和Hisat2 v2.1.0(Kim et al., 2015)分别映射Illumina、PacBio和RNA-seq数据,全面评估了组装质量。同时,还使用基准普遍单拷贝直系同源基因(BUSCO, v2.0.1; Simao et al., 2015)评估了基因组的完整性。
3 结果
3.1 倍性确定、基因型鉴定和基因组大小估计
为了创建适合基因组测序的植物,并且更易于转化和再生,以便在制作转基因植物时提高转化率和再生能力,我们从毛白杨的花药愈伤组织(使用一个其他表现出低转化效率的优良雄性克隆LM50)再生了植株。尽管来源于花药培养,再生植株(GM15;图1a)的倍性水平通过以下多种方法确定。流式细胞仪显示GM15和LM50都是二倍体(图1b)。这一点通过染色体计数进一步得到确认(图1c),并且基于19个等位基因特异性引物,它们的基因型看起来是相同的且为杂合体(表S1;图1d)。此外,GM15和LM50的k-mer频率分布几乎相同,表明基因组大小约为800 Mb,符合二倍体杨树的预期(图1e)。因此,我们得出结论,用于测序的花药再生克隆GM15是从花药中的体细胞而非配子发育而来;因此,它是其亲本毛白杨基因型LM50的合法代表。
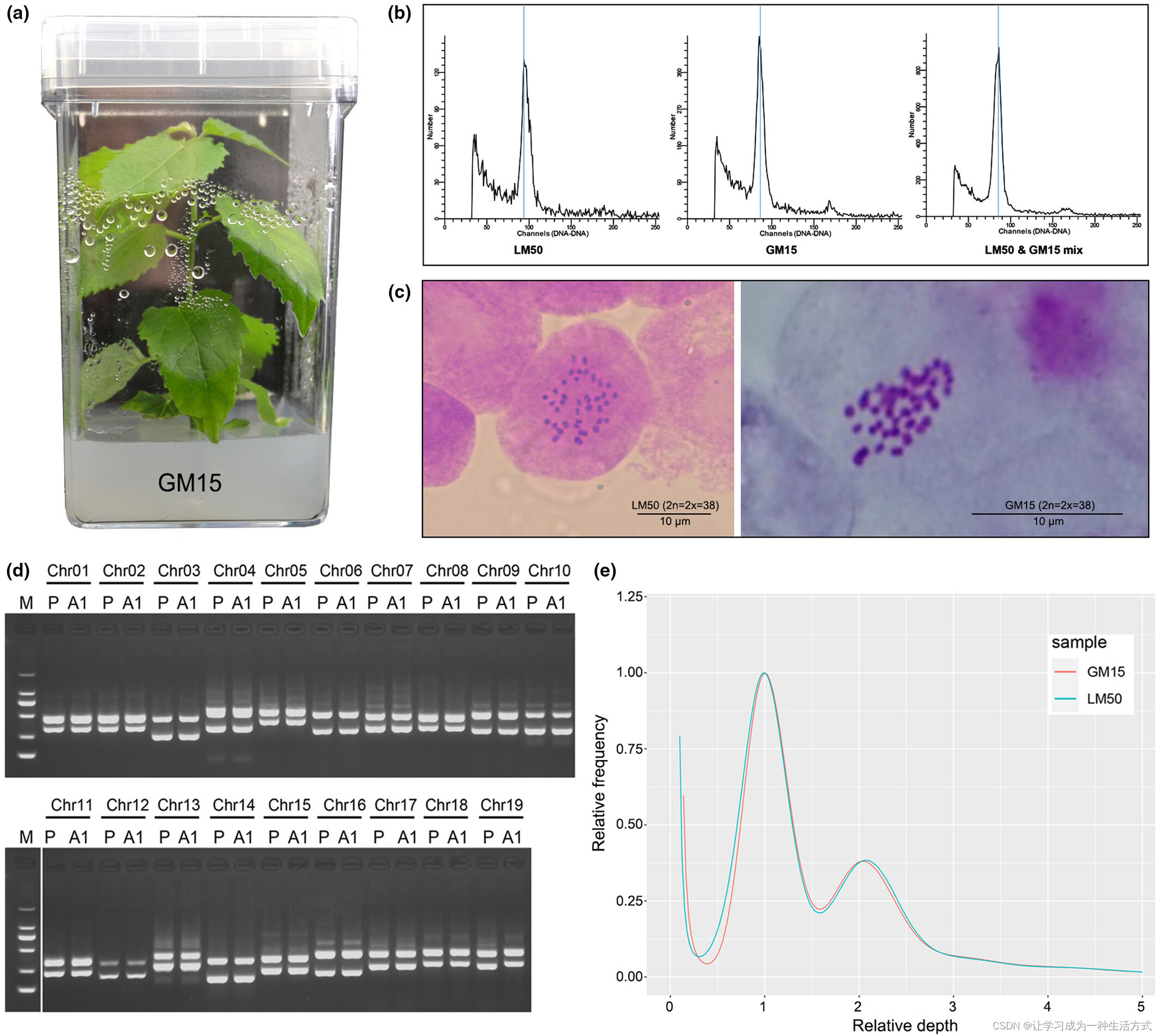
倍性和基因型鉴定,父本LM50及其花药植株GM15 (a)从毛白杨雄性克隆LM50的花药再生的个体(GM15)。(b)通过流式细胞仪检测LM50和GM15的倍性。(c)计数LM50和GM15的染色体。(d)使用来源于19条染色体的等位基因特异性引物通过PCR鉴定LM50和GM15的基因型。(M)标记DL2000,(P)父本(雄性克隆LM50),(A1)花药植株GM15。(e)LM50和GM15的标准化k-mer频率分布。
3.2 基因组组装和染色体锚定
为了获得毛白杨的高质量参考基因组,我们结合使用PacBio、Hi-C和Illumina方法对GM15的基因组进行了测序和组装。通过K-mer分析估计其大小约为800 Mb(图1e,表S3)。过滤后的约54 Gb PacBio数据(6.24百万subreads,约70倍覆盖度)被组装生成初步草稿组装。为了获得染色体规模的组装,Hi-C读数(4.3亿读数,65 Gb,约80倍覆盖度)被测序,并最终成功锚定了38条染色体规模的拟分子体(图2,表S4),生成了740.2 Mb的二倍体基因组大小。38条染色体规模的拟分子体覆盖了估计的800 Mb基因组的92.1%(表1)。Contig N50和scaffold N50的大小分别达到了0.96 Mb和17.13 Mb,最长的contig和scaffold分别为5.47 Mb和46.68 Mb(表1)。
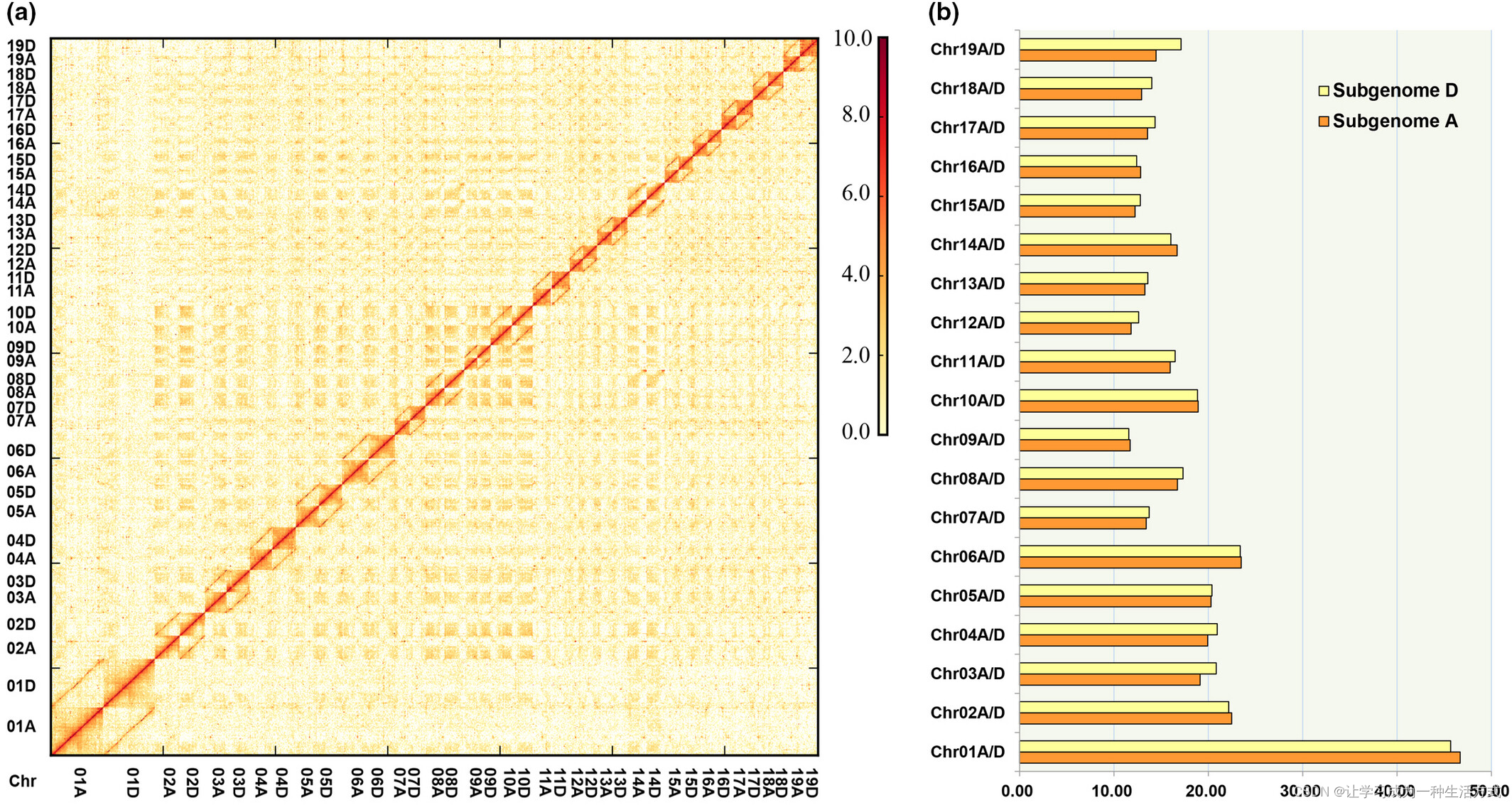
基于染色体规模组装的Hi-C互作热图。(a) 该图展示了通过将Hi-C数据对齐到染色体规模组装上生成的接触矩阵。(b) 来自3D-DNA流水线的两个亚基因组中每条染色体的长度统计。
| Assembly feature | Subgenome A (P. alba var. pyramidalis) | Subgenome D (P. adenopoda) | Genome of P. tomentosa |
|---|---|---|---|
| Estimated genome size by K-mer | -- | -- | 800 Mb |
| Number of contigs | 802 | 845 | 4,025 |
| Contig N50 (bp) | 994,455 | 968,830 | 964,137 |
| Longest contig (bp) | 3,787,650 | 5,467,932 | 5,467,932 bp |
| Contig N90 (bp) | 251,218 | 233,161 | 82,943 |
| Number of scaffolds | 19 | 19 | 2407 |
| Scaffold N50 (bp) | 18,914,766 | 18,843,764 | 17,128,596 |
| Longest scaffold (bp) | 46,677,810 | 45,691,089 | 46,677,810 |
| Scaffold N90 (bp) | 12,249,758 | 12,631,484 | 11,723,923 |
| Assembly length (bp) | 336,656,027 | 344,390,102 | 740,184,868 |
| GC content (% of genome) | 33.42 | 33.17 | 33.60 |
| Gap number | 783 | 826 | 1618 |
| Assembly (% of genome) | -- | -- | 92.11 |
| Repeat annotation (bp/% of assembly) | |||
| LTR | 54,889,361/16.30 | 58,264,760/16.92 | 129,608,743/17.51 |
| Caulimovirus | 300,287/0.09 | 469786/0.14 | 849,811/0.11 |
| Copia | 13,528,294/4.02 | 13,431,562/3.90 | 29,658,574/4.00 |
| Gypsy | 40,866,300/12.14 | 44,086,909/12.80 | 98,553,170/13.31 |
| LINE | 3,312,508/0.98 | 2,828,020/0.82 | 7,766,907/1.05 |
| SINE | 1,979,481/0.59 | 1,814,936/0.53 | 3,925,279/0.53 |
| DNA | 18,854,707/5.60 | 19,016,245 /5.52 | 40,009,905/5.41 |
| RC/Helitron | 18,559,627/5.51 | 19,083,511/5.54 | 41,759,263/5.64 |
| Unknown | 30,157,396/8.96 | 30,468,761/8.85 | 72,521,254/9.80 |
| Satellite | 319,555/0.09 | 481,846/0.14 | 1,632,425/0.22 |
| Simple repeat | 4,442,224/1.32 | 4,738,265/1.38 | 9,640,201/1.30 |
| Low complexity | 1,092,089/0.32 | 1,136,945/0.33 | 2,331,509/0.31 |
| Total repeats | 133,663,317/39.70 | 137,952,318/40.06 | 310,333,451/41.93 |
| Gene annotation(counts) | |||
| Coding gene | |||
| Coding gene number | 28,512 | 28,605 | 59,124 |
| Coding gene number (AED < 0.5) | 27,532 | 27,604 | 57,015 |
| Average gene region length (bp) | 3,429.49 | 3,417.22 | 3,398.78 |
| Average transcript length (bp) | 1,609.1 | 1,602.21 | 1,596.97 |
| Average CDS length (bp) | 1,322.74 | 1,313.56 | 1,313.07 |
| Average exons per transcript | 5.83 | 5.84 | 5.79 |
| Average exon length (bp) | 276.11 | 274.54 | 275.86 |
| Average intron length (bp) | 75.90 | 78.32 | 83.89 |
| Non-coding gene | 1345 | 1331 | 3170 |
| tRNA number | 308 | 308 | 662 |
| rRNA number | 64 | 61 | 436 |
| Other non-coding gene number | 973 | 962 | 2072 |
| Total gene number | 29,857 | 29,936 | 62,294 |
3.3 基因组质量评估、分类和注释
许多其他指标表明该基因组质量很高。Illumina和PacBio数据分别覆盖了全基因组的99.45%和99.76%。RNA-seq数据的映射率为97.8%。总体上,96.5%的BUSCO基因被表示为完整。重复和单拷贝BUSCO核心基因的覆盖深度相同,显示出预期的泊松分布(图S1),表明重复基因不是由组装冗余产生的。此外,在Hi-C热图中未观察到大规模的开关错误,表明这是一个很好的二倍体解析组装。
基于与其他杨树的系统发育关系,毛白杨基因组成功被划分为两个亚基因组(2 × 19条染色体),大小分别为336.7 Mb和344.4 Mb(表1和S5a,b)。组装中同源区域的映射显示出同源染色体对之间的明显同源性,以及不同染色体之间的广泛同源性,这符合高度重复的杨树基因组的预期(图3)。此外,两个亚基因组的覆盖深度由PacBio和Illumina读数均匀覆盖,表明这是一个准确的二倍体解析组装(图S2)。与以前的杨树基因组组装(Ma等,2013a; 2013b; Tuskan等,2006; Yang等,2017)相比,综合评估的结果显示,本研究中毛白杨的组装质量有了显著提高(表S6)。
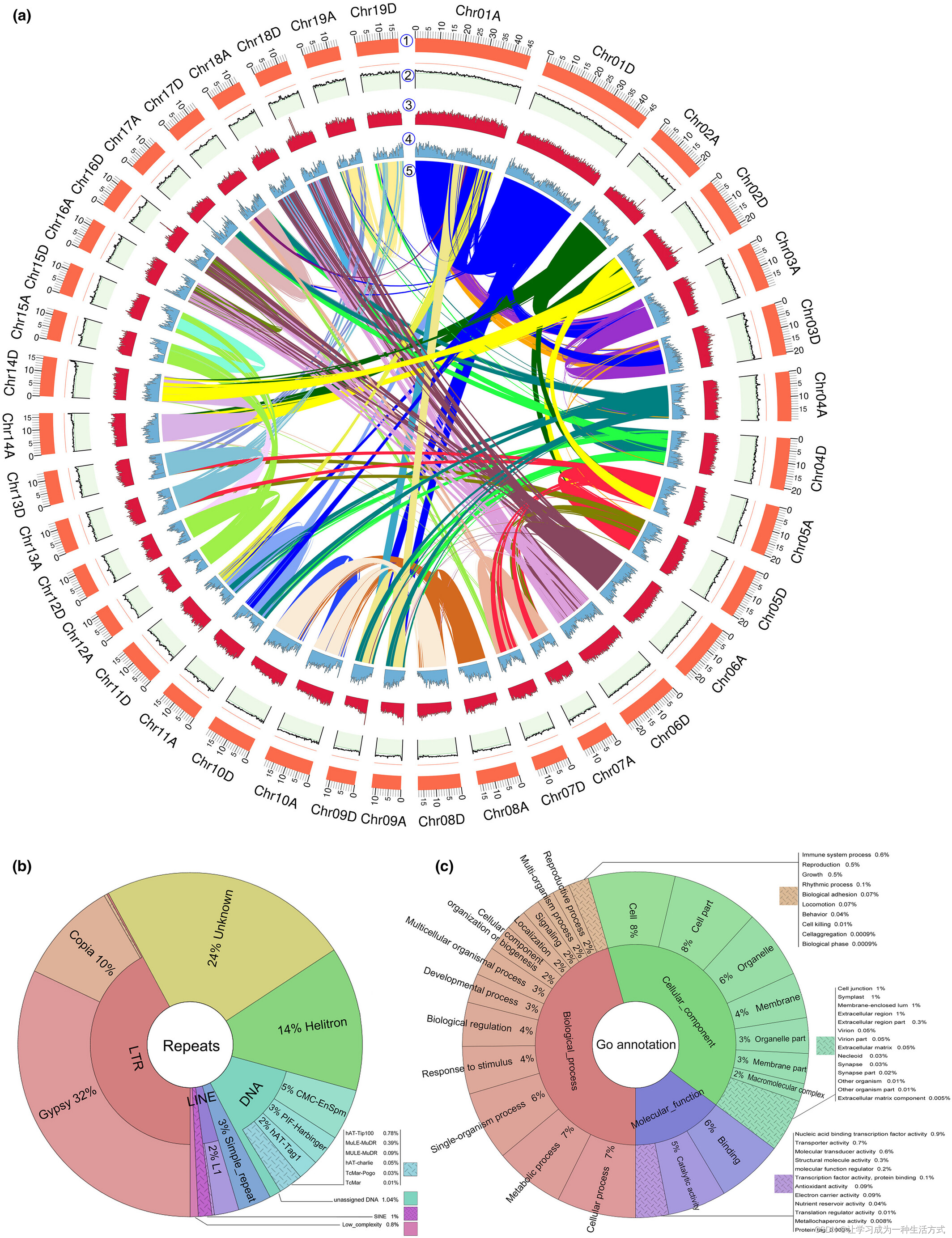
基因组特征描述 (a) 毛白杨基因组概览。在38条染色体上,每200 kb间隔显示的基因组特征。圆周上的单位显示兆碱基值和染色体。① 染色体核型。② GC含量(33.6%,红线代表50%,绿线代表30%)。③ 重复覆盖(45-1,937重复)。④ 基因密度(3-164个基因)。⑤ 同源同源区块。(b) 毛白杨基因组中重复类别的分布。(c) 不同高级基因本体(GO)生物过程术语中预测基因的分布。
总共鉴定出1,001,718个重复序列,这些重复序列的大小为307.6 Mb,约占基因组的41.6%(图3a)。长末端重复(LTR)是最丰富的,占基因组的17.5%。其中13.3%是LTR/Gypsy元素,4.0%是LTR/Copia重复。其次是未知元素,占基因组的9.8%。然后是5.6%的Helitron重复和5.4%的DNA元素(图3b;详见表S7)。两个亚基因组之间重复大小的差异很小;总重复大小分别为亚基因组A和D的133.7 Mb和137.9 Mb(进一步讨论如下),LTR的大小分别为54.9 Mb和58.3 Mb(表1)。
转座元素(TE)的丰度和分布在亚基因组之间有显著差异(图S3)。所有TE类别广泛分布在两个亚基因组上(图S3 ①–⑦)。例如,在亚基因组A和D中分别发现了109,542和114,615个I类TE(增加4.6%),以及101,190和97,478个II类TE(减少3.6%)(图S4)。进一步的皮尔逊卡方检验显示,在Bonferonni校正后(α = 0.0071; 表S8),除LINEs外所有TE类别都有显著差异。
在屏蔽重复序列的毛白杨基因组的基础上,我们首先通过结合73,919个同源蛋白序列和137,918个从毛白杨RNA-seq数据组装的转录本来注释蛋白编码基因。共注释了59,124个蛋白编码基因模型,平均编码序列长度为1.31 kb,每个基因平均6.04个外显子,每个蛋白430个氨基酸。基因区域覆盖了基因组的28.5%,总长度为210.8 Mb(表1,S9,S10)。对蛋白质组的BUSCO评估显示95.8%的完整性,约90%的蛋白序列由已知的蛋白和转录本完整支持,表明基因注释的高质量。然后将注释的基因与三个本体类别相关联:生物过程、细胞组分和分子功能,所有领先的GO术语在第二级可以分类为24个组(图3c)。与蛋白数据库的最大功能注释比率为98.6%(表S11)。我们预测了662个tRNA,总长度为49,659 bp(每个tRNA平均长度为75 bp),以及436个rRNA(106个28S rRNA,106个18S rRNA和224个5S rRNA),长度为610,293 bp。我们还注释了2,072个其他非编码RNA,总长度为218,117 bp。
3.4 比较基因组学和进化
我们将毛白杨基因组中的19,594个基因家族(包含59,124个基因)与其他三个已测序的杨树基因组(包括P. trichocarpa、P. euphratica和P. pruinosa)进行了比较。共鉴定出22,386个基因家族(142,738个基因)。此外,14,738个基因家族(119,375个基因)由所有四个杨树物种共享,1,154个基因家族包含2,038个基因被发现是毛白杨独有的,基于OrthoMCL“相互优化”(图4a,表S12)。
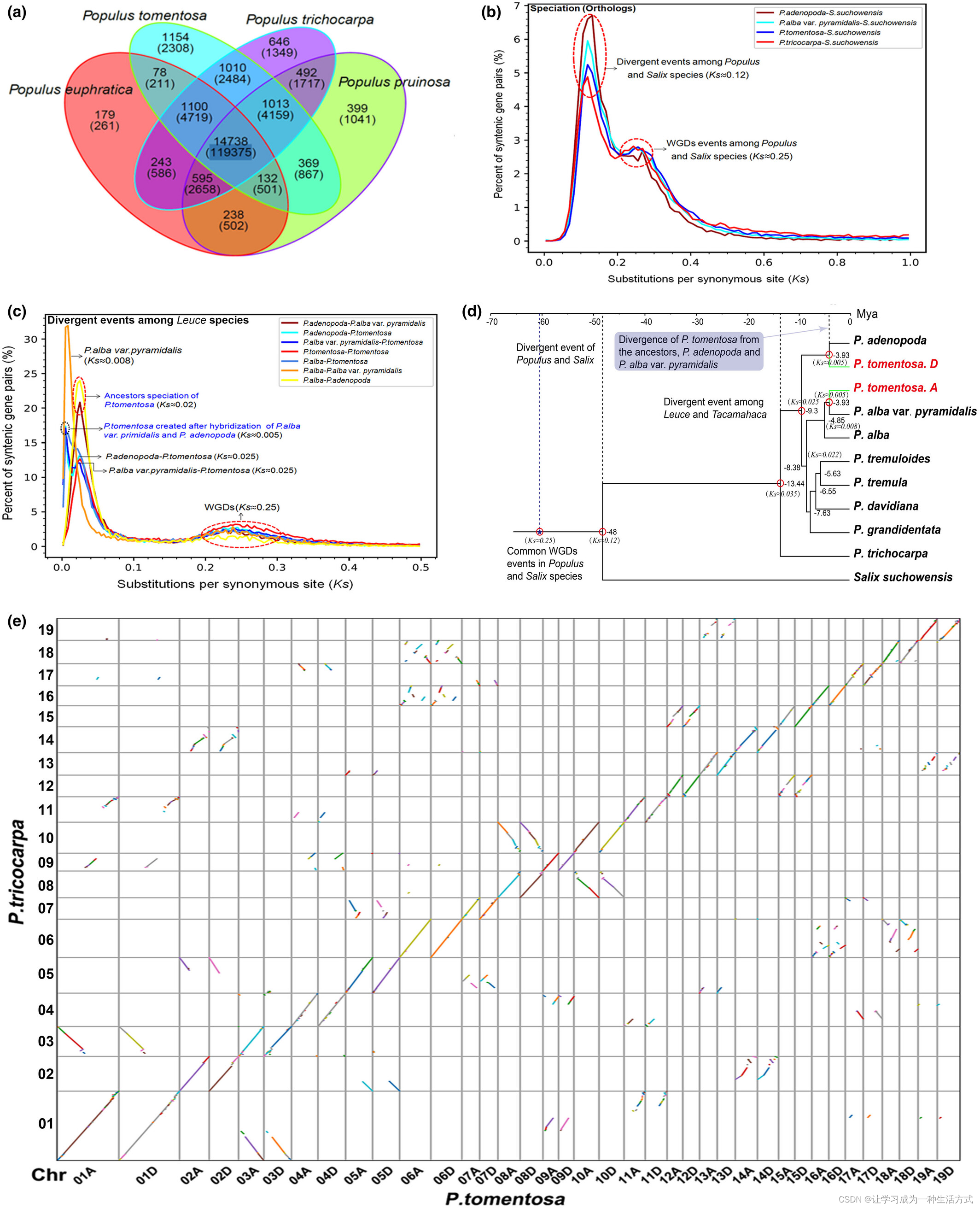
比较基因组学分析 (a) 毛白杨与其他三种杨树共享的基因家族。数字表示每个类别中的家族和基因数量。 (b) 杨柳科物种间和杨树种内通过同源和旁系配对计算的同义替换率(Ks)推断的物种分化。 (c) 杨树和柳树种共有的基因组复制事件(Ks = 0.25,约60 Ma),毛白杨物种形成(Ks = 0.005,约3.93 Ma)以及通过Ks分析揭示的其他杨树的分化事件。 (d) 使用r8s(Sanderson, 2003)推断的10种植物物种的系统发生树。杨柳科物种的全基因组复制(WGD)事件已标记。 (e) 毛白杨基因组(横轴)与P. trichocarpa基因组(纵轴)之间的同源性。基于OrthoMCL分析的同源基因,推断毛白杨染色体与P. trichocarpa染色体具有同源性。
为了确定染色体对并研究毛白杨的亲本起源,我们选择了1052个在每对毛白杨染色体间看似等位的同源基因,这些基因在其他杨树类中是单拷贝基因,然后构建基因树以评估系统发生距离。每对毛白杨染色体的等位基因对在大多数基因树上清晰地最接近P. alba var. pyramidalis(PA)或P. adenopoda(PD)。因此,基于系统发生距离,成功地将毛白杨的总共38条染色体分为两个亚基因组(2×19条染色体)。为了确认结果,我们使用5345个单拷贝同源基因测量了毛白杨两个亚基因组、P. alba var. pyramidalis和P. adenopoda之间的Ks距离。结果也是一致的;我们将毛白杨的基因组称为由P. alba var. pyramidalis衍生的亚基因组A和由P. adenopoda衍生的亚基因组D。
为了探究两个亚基因组之间潜在的重组事件,估计了5345个毛白杨、P. alba var. pyramidalis和P. adenopoda单拷贝同源基因的同义(Ks)距离。我们发现在大多数基因位点(4309:80.62%)内几乎没有明显的重组事件,尽管出现了低水平的重组(38个位点,0.87%);998个位点(18.7%)不符合上述两种假设,因此无信息(表S13,图S5)。这表明毛白杨的两个亲本亚基因组可能在很大程度上仍保持完整,至少在基因组成方面是如此。
我们重建了亚基因组A、亚基因组D和其他杨树的系统发生树(图S6),以及相应的19对染色体的每一对的系统发生树(图S7)。所有这些分析都支持了毛白杨基因组起源于P. adenopoda和P. alba var. pyramidalis之间的杂交的假设。基于P. alba var. pyramidalis是雄性克隆的事实,以及没有发现雌性克隆,结合对杨属的叶绿体和线粒体基因组的系统发生分析(图S8–S9),这表明P. adenopoda是毛白杨的母本亲本,我们推断P. alba var. pyramidalis和P. adenopoda分别是毛白杨杂交形成的父本和母本。
3.5 染色体结构变异和GO分析
为了确定杨树物种分化和复制事件的日期,我们对从Populus物种和Salix suchowensis衍生的同源基因对进行了MCScanX(Wang et al., 2012)的共线性分析。从Ks(同义替换率)分布中,我们推断出一个全基因组复制事件(WGD;基于旁系配对)和一个物种分化事件(基于同源配对)。四种杨树物种和S. suchowensis的同源基因中的Ks分布包含两个峰值。一个峰值表明杨树和柳树物种经历了一个共同的WGD事件(Ks约0.25)。这些WGD事件在被子植物的进化中已知频繁发生(Jiao等,2011; Myburg等,2014; Otto, 2007; Van de Peer等,2017)。这个结果也与之前关于Salix suchowensis的研究(Dai等,2014)一致。另一个代表Populus和Salix之间分化的峰值也可见(Ks约0.12;图4b)。进一步分析显示,杨属和Tacamahaca(P. trichocarpa)分部的分化大约在13.4 Ma发生。毛白杨的一个祖先P. adenopoda首先在大约9.3 Ma时从Populus家族中独立出来形成一个独立的类群。随后,白杨组和白杨组经历了一个分化事件(大约8.4 Ma)。毛白杨的另一个祖先P. alba var. pyramidalis在大约4.8 Ma产生了P. alba的一个独立变种。大约在3.9 Ma,P. tomentosa通过P. adenopoda(女)和P. alba var. Pyramidalis(男)之间的杂交形成(图4d)。进一步使用白杨种和P. trichocapa的叶绿体和线粒体基因组构建的系统发生树支持P. adenopoda最可能是毛白杨的母本(图S8和S9)。
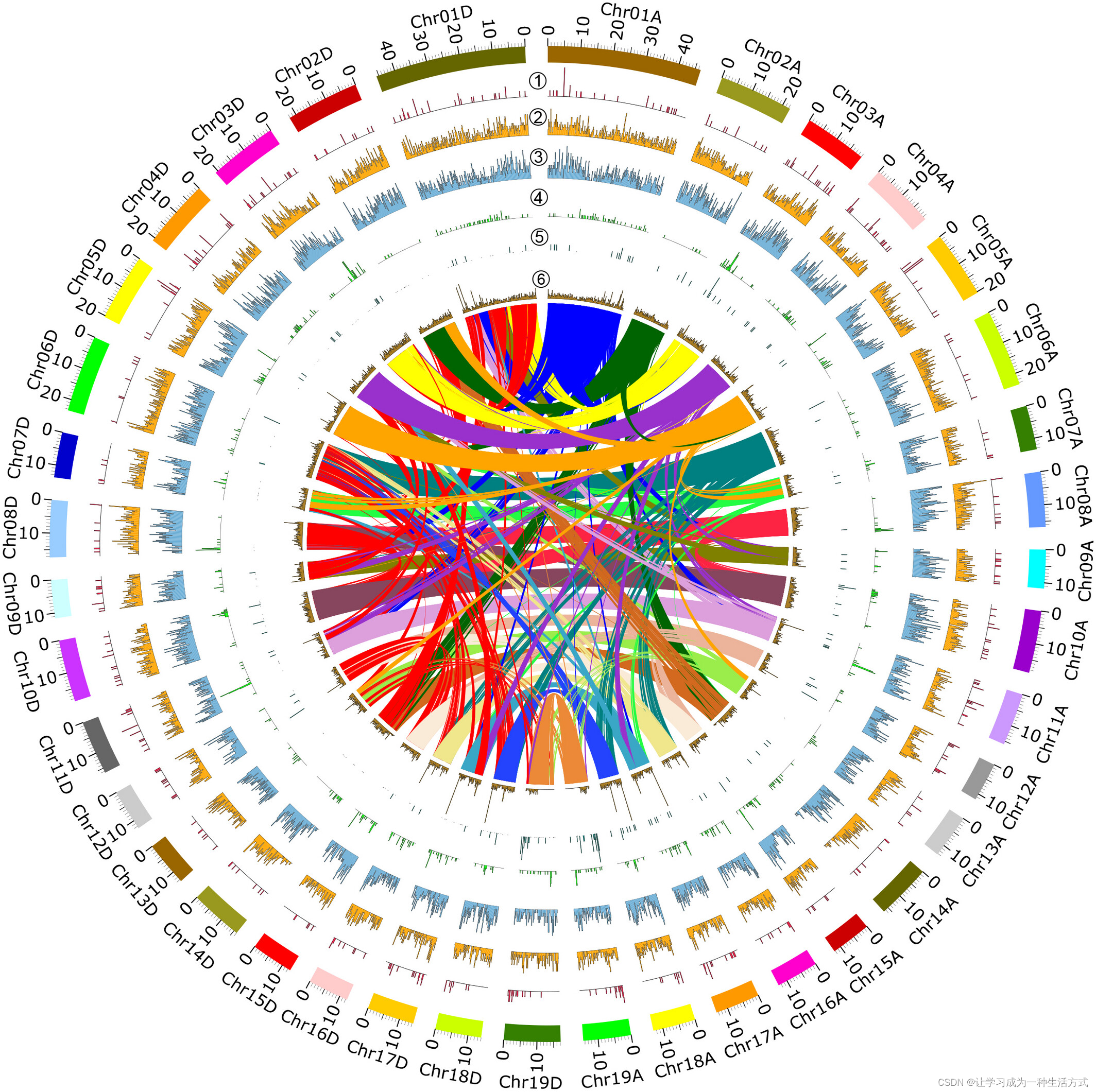
染色体结构变异和等位基因间插删除分析 在毛白杨两个亚基因组之间: ① 染色体核型, ② 拷贝数变异(CNV)的基因组分布, ③ 缺失(DEL)的基因组分布, ④ 插入(INS)的基因组分布, ⑤ 倒位(INV)的基因组分布, ⑥ 易位(TRANS)的基因组分布, ⑦ 两个毛白杨亚基因组的等位基因间的插入/缺失(indels)的基因组分布。 ⑧ 内部部分显示亚基因组A和亚基因组D之间的同源性。基于OrthoMCL分析中识别的同源基因,推断亚基因组A的染色体与亚基因组D的染色体具有同源性。
基因组范围的结构变异(SV),包括拷贝数变异(CNV)、缺失(DEL)、插入(INS)、倒位(INV)和易位(TRANS)等(图5,标为圆圈数字①至⑤),表明毛白杨基因组中存在丰富的染色体结构变异。在整个基因组中,我们共检测到15,480个结构变异,其中INS(6654)和DEL(6231)占大多数(83%)。其他变异数量为1602和694,以及299个分别为INV,TRANS和CNV,共占观察到的SV总数的27%(表S15)。大多数INS、DEL和CNV变异发生在同源染色体对之间,而TRANS通常出现在非同源染色体对之间(表S15,图S10)。我们还发现SV影响了基因结构和氨基酸序列。例如,等位基因对Potom02G0005700和Potom02G0411400的插入/缺失引起了核酸和氨基酸序列的变化(图S11)。
我们观察到总共299个CNV在整个基因组中有不规则和零星的分布(图5)。相对而言,在Chr17A和Chr17D(0.54/Mb)、Chr09A和Chr09D(0.47/Mb)上看到了高密度的CNV,而在Chr06A和Chr06D(0.13/Mb)、Chr13A和Chr13D(0.15/Mb)、Chr07A和Chr07D(0.18/Mb)上分布了相对低密度的CNV(图5②)。我们还注意到,大多数DEL几乎均匀分布在整个基因组中,略微偏好于Chr12A、Chr12D、Chr17A、Chr17D、Chr18A和Chr18D的端粒区域(图5③)。类似地,INS呈现高密度,并稍微偏好于Chr07A、Chr07D、Chr15A、Chr15D、Chr18A和Chr18D的端粒区域(图5④)。与此相反,INV在基因组中的分布更不均匀(图5⑤)。INV在Chr01A和Chr01D上更为丰富,而在其他染色体上的分布有限。TRANS在染色体上的分布非常稀疏,只在Chr02D、Chr07D、Chr08D、Chr13D和Chr14D上检测到少数(图5⑥)。
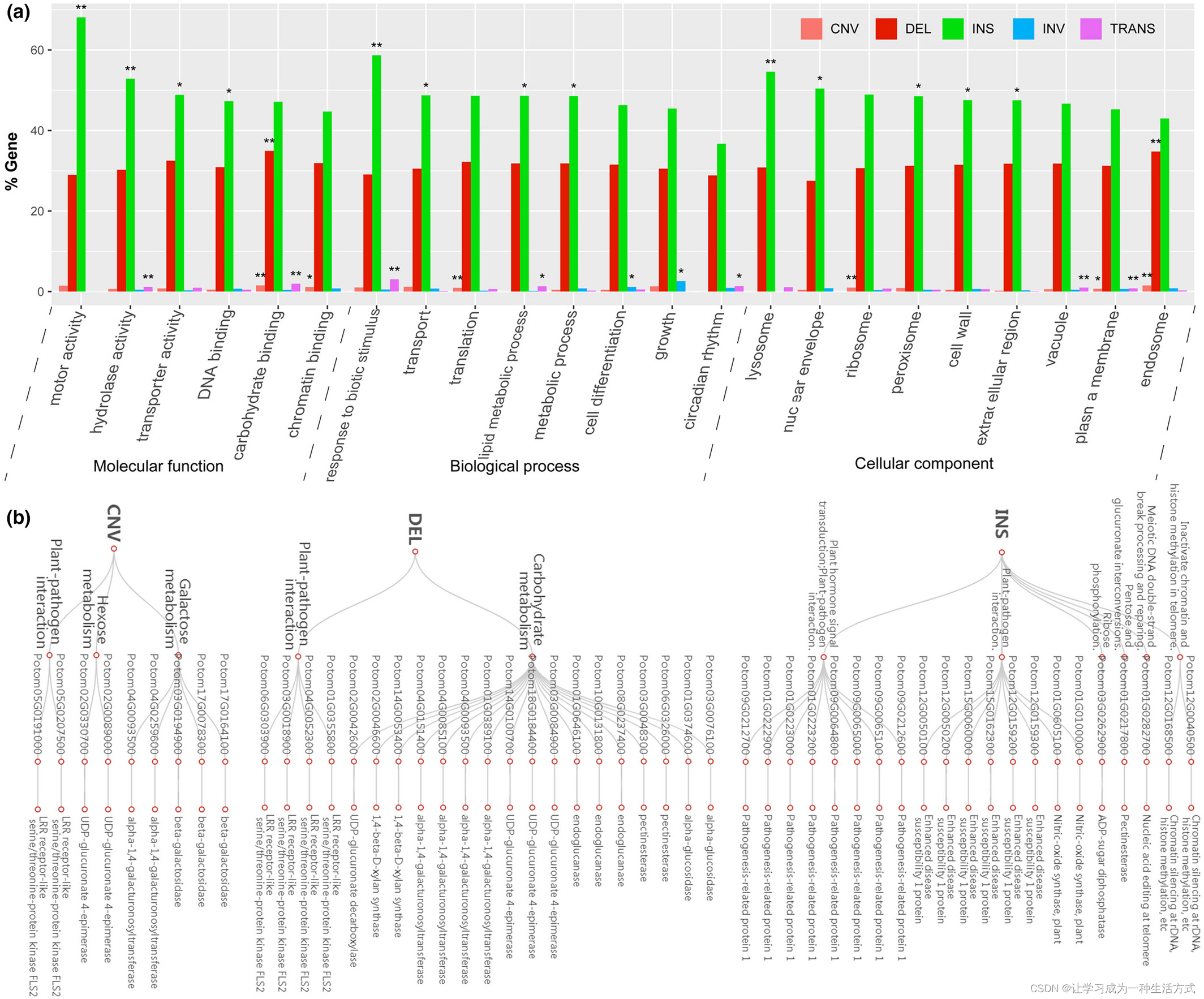
我们对位于总共15,480个SV区域的基因进行了GO富集分析,并检测到23个GO类别显著过表达(图6)。其中十个("motor activity"、"transporter activity"、"DNA binding"、"transport"、"metabolic process"、"lysosome"、"nuclear envelope"、"peroxisome"、"cell wall" 和 "extracellular region")在受INS影响的基因中过表达,三个("chromatin binding"、"translation" 和 "ribosome")在受CNV影响的基因中过表达,三个("hydrolase activity"、"response to biotic stimulus" 和 "lipid metabolic process")在受INS和TRANS影响的基因中过表达,两个("cell differentiation" 和 "growth")在受INV影响的基因中过表达,两个("vacuole" 和 "circadian rhythm")在受TRANS影响的基因中过表达,一个("endosome")在受DEL和CNV影响的基因中过表达,一个("carbohydrate binding")在受DEL、CNV和TRANS影响的基因中过表达,一个("plasma membrane")在受CNV和TRANS影响的基因中过表达。总体而言,功能注释显示与所有主要GO类别相关的富集(图6a)。
染色体结构变异和等位基因插入/缺失(indels)分析 在毛白杨的亚基因组A和亚基因组D之间,我们观察到以下特征: ① 染色体核型, ② 拷贝数变异(CNV)的基因组分布, ③ 缺失(DEL)的基因组分布, ④ 插入(INS)的基因组分布, ⑤ 倒位(INV)的基因组分布, ⑥ 易位(TRANS)的基因组分布, ⑦ 两个毛白杨亚基因组之间的等位基因的indels基因组分布。 ⑧ 亚基因组A和亚基因组D之间的内部部分显示了同源性。基于OrthoMCL分析中识别的同源基因,推断亚基因组A的染色体与亚基因组D的染色体具有同源性。
基因组范围内的结构变异(SV),包括拷贝数变异(CNV)、缺失(DEL)、插入(INS)、倒位(INV)和易位(TRANS)等(图5,圈中数字1-5),表明毛白杨基因组中存在丰富的染色体结构变异。在整个基因组中,我们共检测到15,480个结构变异,其中INS(6654)和DEL(6231)占多数(83%)。其他变异数量分别为1602个INV、694个TRANS和299个CNV,这些变异共占观察到的SV总数的27%(表S15)。大多数INS、DEL和CNV变异发生在同源染色体对之间,而TRANS通常见于非同源染色体对之间(表S15,图S10)。我们还发现SV影响了基因结构和氨基酸序列。例如,带有CNV的Potom05G0191000和Potom05G0207500,带有DEL的Potom06G0303900和Potom01G0355800基因,它们都编码LRR受体样丝氨酸/苏氨酸蛋白激酶FLS2,这可能对疾病抗性很重要。在INS区域的疾病抗性基因主要注释为一氧化氮合酶、增强病害敏感性1蛋白和与植物激素信号转导及植物-病原体互作相关的致病相关蛋白1。更有趣的是,我们在亚基因组P. adenopoda中发现Potom05G0191000和Potom05G0207500各有3个拷贝,在亚基因组P. alba var. pyramidalis中各有11个拷贝。先前在大豆(Glycine max, McHale et al., 2012)的研究也表明,与疾病抗性和生物压力相关的基因中常见结构变异如CNV。Potom05G0191000和Potom05G0207500的更多拷贝数可能有助于解释为什么优良个体LM50表现出强烈的疾病抗性——这是森林种植者所熟知的特征。当然,这个假设需要功能验证。
我们还发现许多参与碳水化合物代谢的基因存在包括CNV、DEL和INS在内的结构变异。例如,UDP-葡萄糖醛酸4-表酶、alpha-1,4-半乳糖醛酸基转移酶和beta-半乳糖苷酶(图6b)。此外,显示INS变异的Potom03G0262900和Potom01G0217800被注释为ADP糖二磷酸酶和果胶酯酶,并参与核糖磷酸化和戊糖及葡萄糖醛酸转化;它们可能对能量和生长很重要。最后,众所周知,着丝粒和端粒在维持染色体稳定性中起着重要作用。有趣的是,我们还发现三个基因Potom01G0282700、Potom12G0168500和Potom12G0040500显示了INS变异,它们参与减数分裂DNA断裂处理和修复、rDNA的染色质沉默和组蛋白甲基化。这些基因可能在维持染色体结构或减少我们观察到的减数分裂重组率中发挥作用。
4 讨论
尽管单倍体诱导并不成功,但我们获得了一个更加鲜嫩、易于再生和可转化的个体GM15,该个体在染色体组、基因型和基因组大小等证据上与其亲本树木LM50非常相似,因此被认为适合进行测序。在这里,我们整合了先进的SMRT测序技术(PacBio)、Illumina校正和染色体构象捕获(Hi-C)来组装一个高质量的单倍型解析基因组。与几个已发表的杨树基因组相比,包括P. trichocarpa(Tuskan等人,2006)、P. euphratica(马等人,2013a;2013b)、P. pruinosa(杨等人,2017)和P. alba var. pyramidalis(马等人,2019),毛白杨的组装质量更高或具有可比较的质量。只有P. alba基因组的contig N50比毛白杨长(1.18与0.96 Mb);然而,其contigs尚未与特定染色体相关联(刘等人,2019;表S7)。毛白杨的整个基因组大小为740.2 Mb,由亚基因组A(P. alba var. pyramidalis)和亚基因组D(P. adenopoda)的总和组成。它与P. trichocarpa(422.9 Mb)、P. euphratica(497.0 Mb)和P. pruinosa(479.3 Mb)的基因组大小明显不同,以及P. alba var. pyramidalis(464.0 Mb)和P. alba(416.0 Mb),这些基因组分别由19条染色体组成,因为这些二倍体的等位基因多样性被归并为单倍体基因组而不是两个二倍体亚基因组(刘等人,2019;马等人,2013a;2013b;Tuskan等人,2006;杨等人,2017)。然而,这种情况与最近发表的一个杂交杨树(84 K)的基因组非常相似,它被划分为两个亚基因组(P. alba和P. tremula var. glandulosa),总基因组大小为747.5 Mb(邱等人,2019;表S7)。
我们提出了杨树属内部及P. tomentosa谱系内的分歧和重复事件的证据。与其他许多开花植物一样(Otto,2007),杨柳科物种在Salix和Populus分化之前经历了一个共同的古六倍体事件,随后是一个古四倍体事件(Lin等人,2018;刘等人,2019;Tuskan等人,2006)。随后,杨树的物种分化逐渐发生。毛白杨、毛白杨和P. alba var. pyramidalis的祖先大约在约930万年和480万年前从Populus组中逐渐分化出来。毛白杨大约在390万年前从杂交事件中出现。这一发现与先前关于毛白杨起源的提议不同(王等人,2014)。与大多数其他测序的杨树不同(马等人,2013a;2013b;Tuskan等人,2006;杨等人,2017),毛白杨基因组由亚基因组A(P. alba var. pyramidalis)和亚基因组D(P. adenopoda)组成(图3和表1)。在毛白杨内部似乎也存在与其杂交起源相关的变异。根据少量标记基因的研究,王等人(2019)提出,毛白杨的雄性亲本物种可能是白杨,但母本可能是P. adenopoda或P. davidiana(对应于毛白杨类型mb1和mb2,分别为P. tomentosa的类型mb1和mb2;王等人,2019)。然而,山东原产地的毛白杨未被收集到他们的实验材料中,非常巧合的是,我们研究中的精英毛白杨克隆体LM50正是来自山东原产地,与毛白杨类型mb1和mb2有所不同。因此,毛白杨可能有比完全理解的更复杂的进化历史,可能包括多次独立的起源。
我们对基因内重组事件的分析表明,尽管两个P. tomentosa亚基因组共享了约393万年的同一核,但它们在很大程度上保持了独立。为了评估自物种起源以来这种低重组率是否符合预期,我们使用了最近一项针对紧密相关的欧洲白杨(P. tremula)的重组数据(假设这种非杂交物种显示出普通的杨树重组率,以生成预期的重组率)。他们估计重组率为15.6–16.1 cM/Mbp/代(Apuli等人,2020)。一般来说,毛白杨的生命周期很长,幼苗在至少7-8年后开始开花,之后在生殖期间每年都会开花(朱,1992)。假设一代时间约为20年,则预计每1 kb基因会有31次重组,比我们的观察结果低几个数量级。这表明,尽管在研究中的基因内发生了重组事件的充足机会,但P. tomentosa的两个亚基因组在许多千代以来基本上保持完整。P. tomentosa的亚基因组完整性,其中似乎存在低正常减数分裂产物率,与该物种非常低的育性观察结果一致。在对毛白杨的精英树木资源进行的研究中,大多数都显示出较低的育性,种子结实率低,发芽率低,幼苗存活率低(白,2015)。这样的特征以及对毛白杨的最近遗传分析(王等人,2019)表明,毛白杨表现出了类似广杂交的F1代,具有相当有限但不为零的育性。
在真核生物中,SVs越来越被认为是导致表型变异的主要因素(Gabur等人,2019)。在植物中,SVs已被证明与诸如植物高度(周等人,2015)和生物胁迫抗性(Cook等人,2012)等许多表型变异密切相关。在我们的研究中,我们检测到GM15基因组中的15,480个SVs,其中12,885个是INDELS,占SVs的大多数(83%)。GO分析表明INDELS在植物-病原体相互作用和碳水化合物代谢等与基因功能密切相关的基因中高度表达。因此,它们可能对毛白杨的疾病抗性和快速生长等特性有所贡献。一些INDELS也富集在与减数分裂DNA双链断裂处理和修复,以及端粒染色质和组蛋白甲基化失活相关的基因中。也许这些SVs有助于保持两个亚基因组的独立性并维持毛白杨的染色体型稳定性,从而在维持其假设的“固定杂种优势”方面发挥作用,下文将进一步讨论。我们还发现了299个CNVs,GO分析表明与植物激素信号转导、植物-病原体相互作用和糖代谢有关。总之,毛白杨中发现的许多SVs为研究其生物学作用和表型效应提供了合理的研究焦点,与杂种优势、进化、育种和生物技术相关。
亚基因组之间低重组的机制尚不清楚。毛白杨因其低性育性而闻名(马等人,2013a;2013b),这可能反映了导致异常配子的减数分裂困难。正如对南瓜亚基因组所提出的建议(Sun等人,2017),毛白杨基因组中的低重组率可能是由于两个亲本物种之间的重复DNA组成迅速分化,这可能抑制了同源染色体的减数分裂配对和后续的交换;如上所示,两个基因组的转座子组成存在显着差异。此外,转座子活性可能会导致CNVs、INSs、TRANSs和DELs,因为它们能够在染色体内和染色体之间动员和重组基因序列(Morgante等人,2007),无论是在野外还是在育种过程中(Lisch,2013)。这些SVs可能进一步抑制正常的减数分裂。在古代异源四倍体南瓜基因组(Sun等人,2017)和新合成的异源四倍体小麦基因组中观察到了染色体型稳定性和亚基因组之间罕见的重组。然而,它们与抑制重组率的功能联系尚不清楚。我们发现的毛白杨亚基因组的保持可能具有提供一定程度的“固定杂种优势”的优势。这可能有助于解释毛白杨尽管性育性低但产量高、分布广泛的特点。

















 1723
1723

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









