









一、引言
ggpicrust2包由凯斯西储大学张亮亮团队发布。ggpicrust2是一个综合的 R 包,旨在为分析和解释PICRUSt2功能预测结果。它提供了多项功能,包括路径名称/描述注释、高级差异丰度(DA)分析方法,以及 DA 结果的可视化,帮助研究人员深入理解微生物群落的功能潜力。该方法自发布以来广受重视和使用。
该包的独特功能之一是能够比较不同 DA 方法在同一数据集上的一致性和不一致性。允许用户在预测和测序特定样本的宏基因组时评估各种方法之间的一致性和差异性,评估结果的稳健性。通过对不同方法进行比较,他们能够根据对同一样本的宏基因组预测或测序结果的一致性评估做出明智的决定并得出可靠的结论。如下所示,是ggpicrust2的工作流程,可根据自己数据的类型选择响应的工作流程。
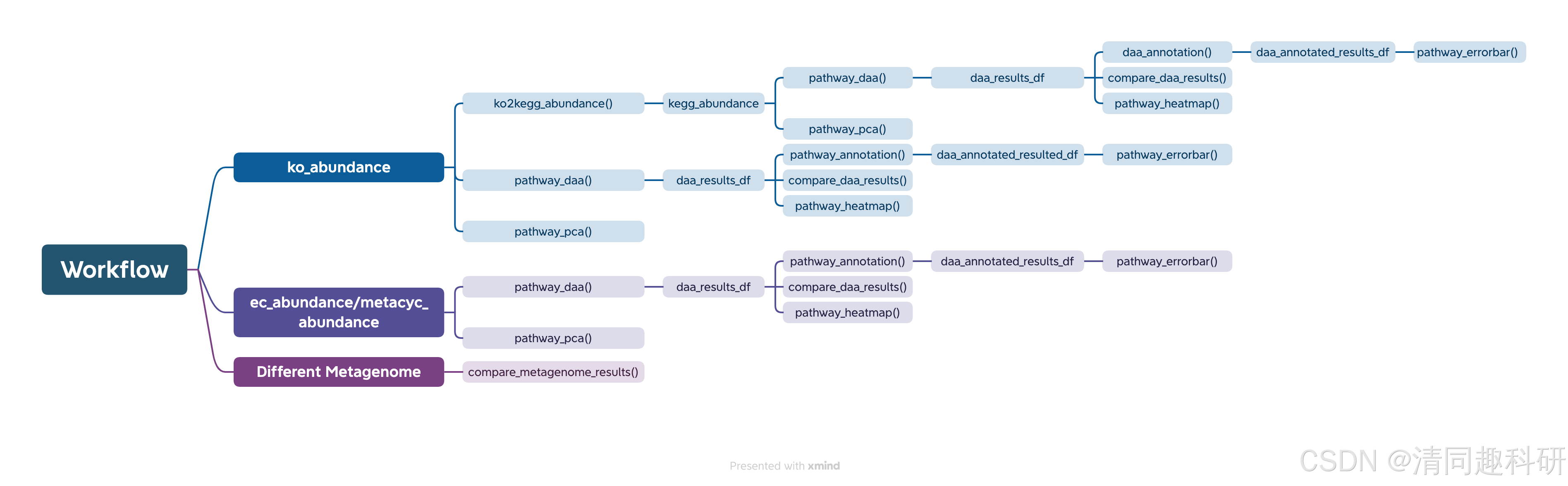
二、文献美图欣赏
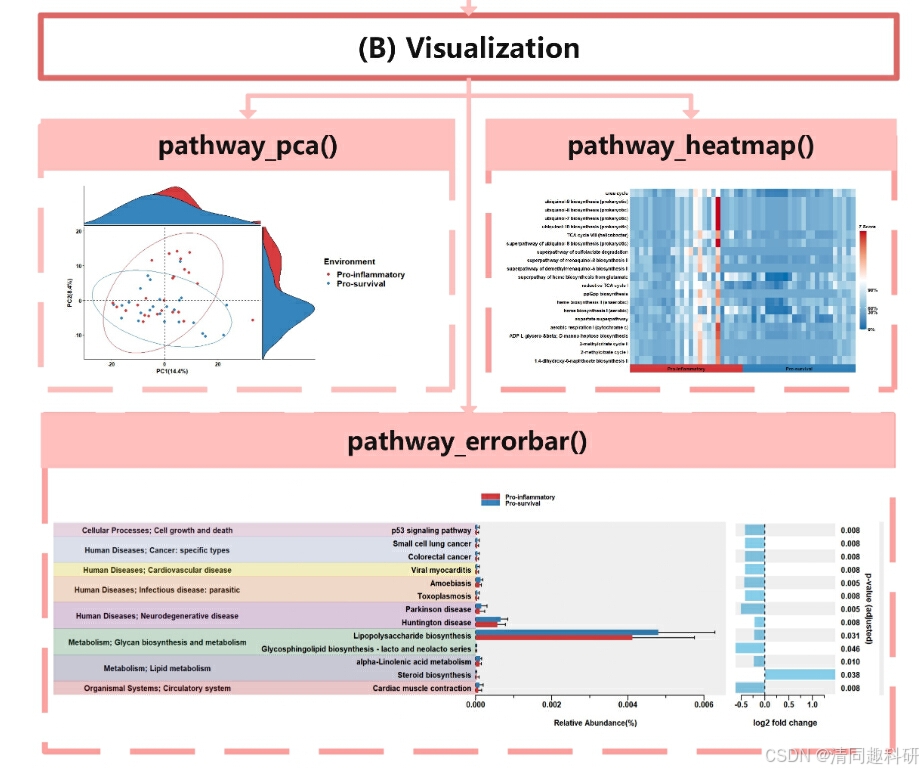
ggpicrust2原文中提供的可视化图片。
三、实例数据和R代码
🌟安装并加载ggpicrust2包
ggpicrust2依赖众多的CRAN和Bioconductor包,因此使用的时候要确包众多依赖包安装完全。
#####################ggpicrust2包安装#############################
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
pkgs <- c("phyloseq", "ALDEx2", "SummarizedExperiment", "Biobase", "devtools",
"ComplexHeatmap", "BiocGenerics", "BiocManager", "metagenomeSeq",
"Maaslin2", "edgeR", "lefser", "limma", "KEGGREST", "DESeq2")
for (pkg in pkgs) {
if (!requireNamespace(pkg, quietly = TRUE))
BiocManager::install(pkg)
}
#从CRAN中安装稳定版ggpicrust2包
if(!require("ggpicrust2")) {install.packages("ggpicrust2")}
#要从 GitHub 安装ggpicrust2的最新开发版本
#if(!require("devtools")){install.packages("devtools")}
#devtools::install_github("cafferychen777/ggpicrust2")
🌟使用包里自带的数据运行ggpicrust2包。
# 加载示例数据
data(ko_abundance)
data(metadata)
# 进行ggpicrust2分析
results_file_input <- ggpicrust2(data = ko_abundance,
metadata = metadata,
group = "Environment",
pathway = "KO",
daa_method = "LinDA",
ko_to_kegg = TRUE,
order = "pathway_class",
p_values_bar = TRUE,
x_lab = "pathway_name")
results_file_input
# 保存图像
ggsave('pic1.png', width = 15, height = 5, bg = 'white')
🌟输出结果
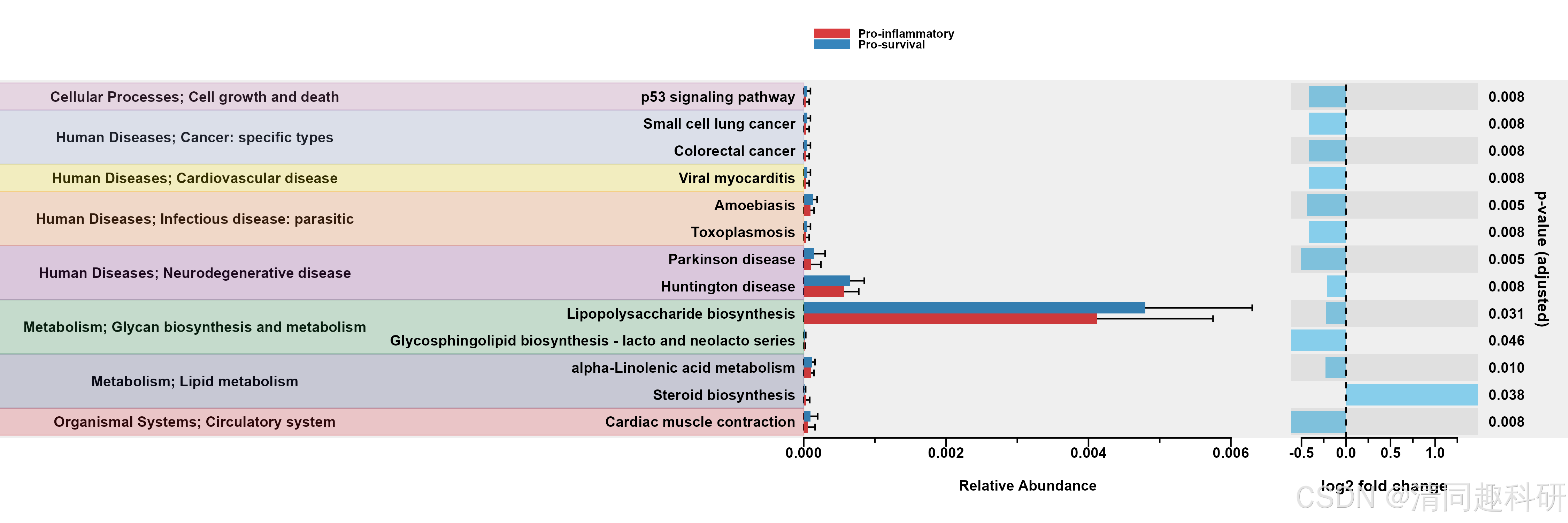
🌟通路热图分析
##################通路热图########################
library(tidyverse)
library(ggh4x)
library(ggpicrust2)
# 加载数据
data("metacyc_abundance")
data("metadata")
# 进行差异表达分析
metacyc_daa_results_df <- pathway_daa(
abundance = metacyc_abundance %>% column_to_rownames("pathway"),
metadata = metadata,
group = "Environment",
daa_method = "LinDA"
)
# 注释结果
annotated_metacyc_daa_results_df <- pathway_annotation(
pathway = "MetaCyc",
daa_results_df = metacyc_daa_results_df,
ko_to_kegg = FALSE
)
# 使用 p < 0.05过滤特征值
feature_with_p_0.05 <- metacyc_daa_results_df %>%
filter(p_adjust < 0.05)
# 绘制热图
pathway_heatmap(
abundance = metacyc_abundance %>%
right_join(
annotated_metacyc_daa_results_df %>% select(all_of(c("feature","description"))),
by = c("pathway" = "feature")
) %>%
filter(pathway %in% feature_with_p_0.05$feature) %>%
select(-"pathway") %>%
column_to_rownames("description"),
metadata = metadata,
group = "Environment"
)
🌟输出结果
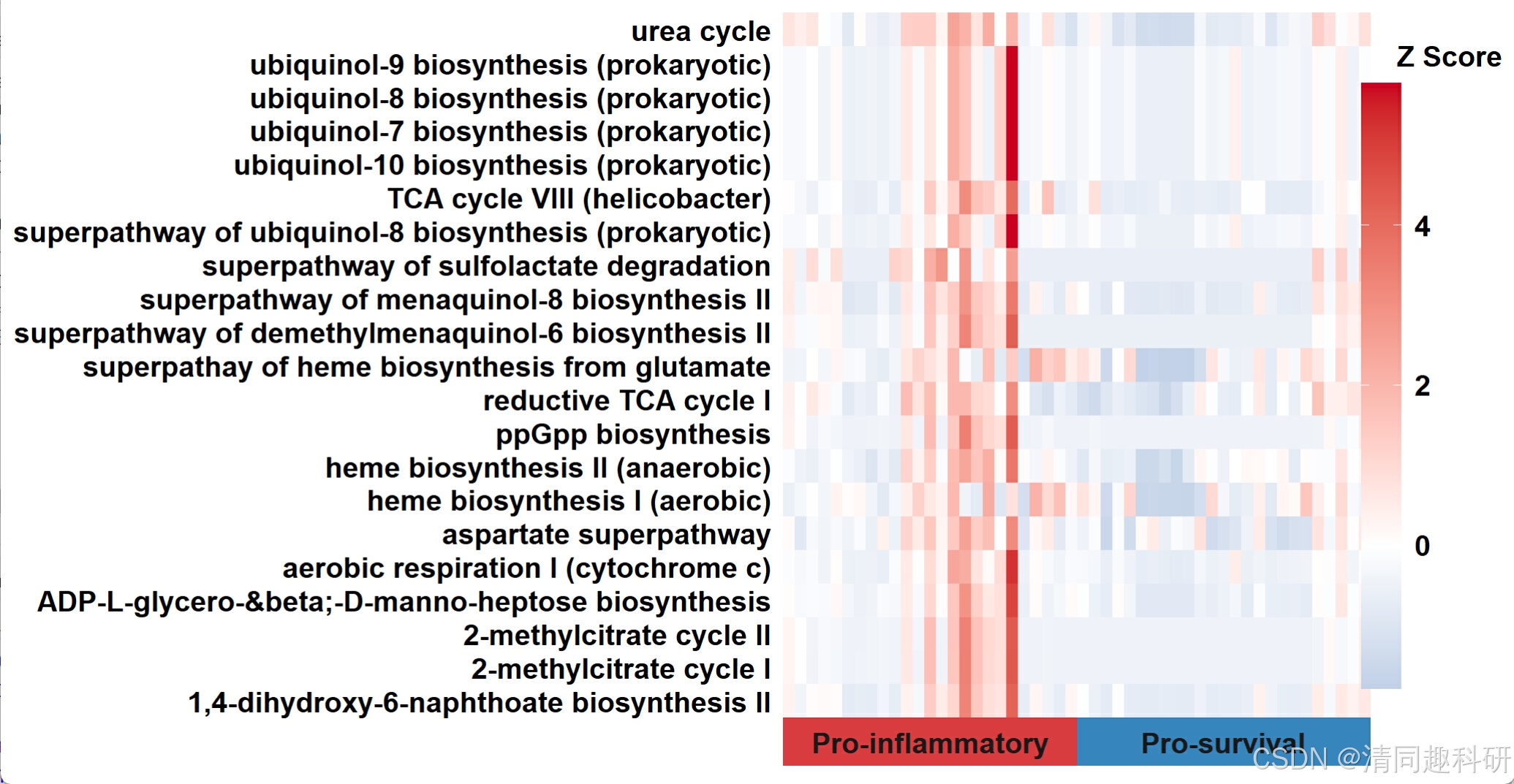
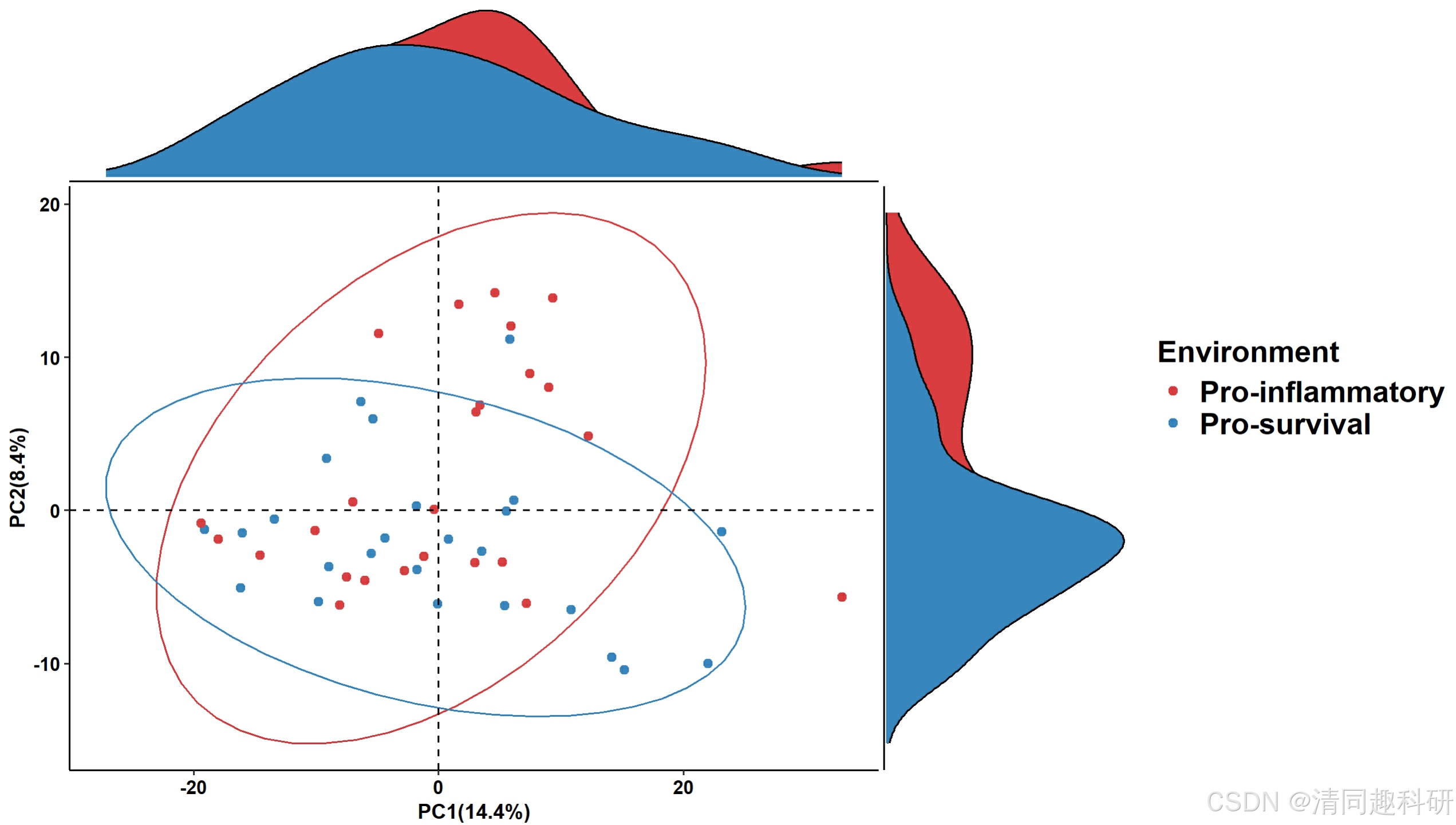
🌟通路PCA分析
##################通路PCA###########################
# Create example functional pathway abundance data
data("metacyc_abundance")
data("metadata")
pathway_pca(abundance = metacyc_abundance %>% column_to_rownames("pathway"), metadata = metadata, group = "Environment")
🌟输出结果
四、参考文献
Chen Yang, Jiahao Mai, Xuan Cao, Aaron Burberry, Fabio Cominelli, Liangliang Zhang, ggpicrust2: an R package for PICRUSt2 predicted functional profile analysis and visualization, Bioinformatics, Volume 39, Issue 8, August 2023, btad470
五、相关信息
!!!本文内容由小编总结互联网和文献内容总结整理,如若侵权,联系立即删除!
!!!有需要的小伙伴评论区获取今天的测试代码和实例数据。
📌示例代码中提供了数据和代码,小编已经测试,可直接运行。
以上就是本节全部结果。
如果这篇文章对您有用,请帮忙一键三连(点赞、收藏、评论、分享),让该文章帮助到更多的小伙伴。

















 4730
4730

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









