






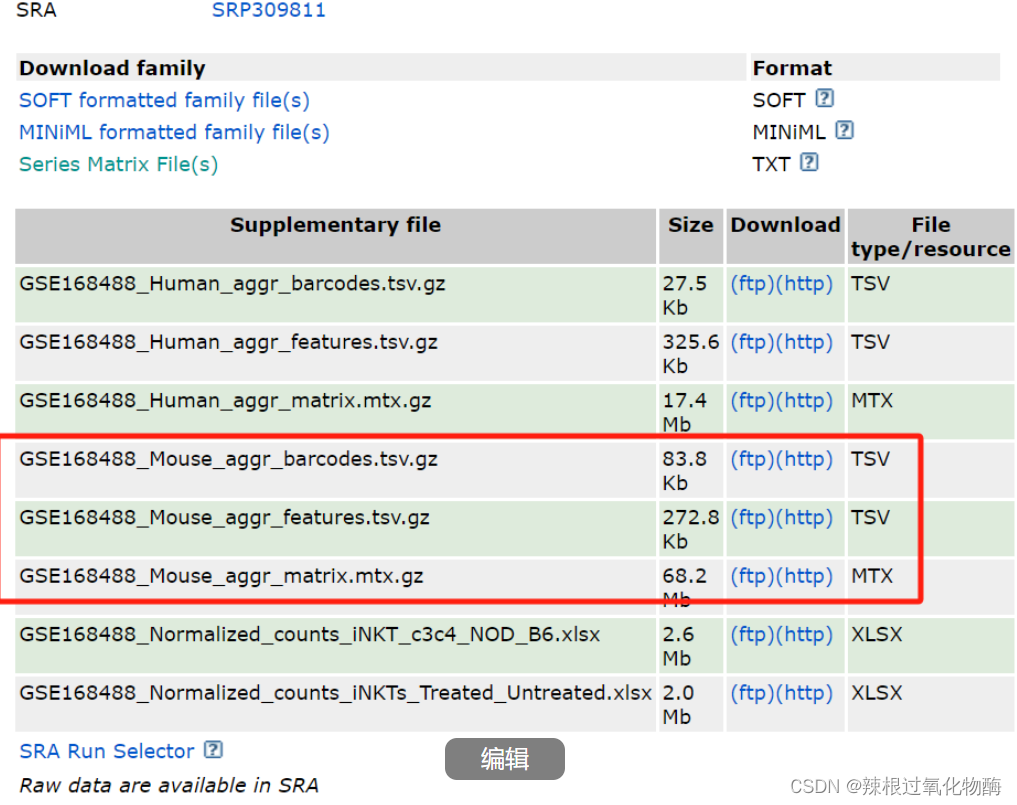
分析GSE168488的小鼠10X单细胞数据时,发现作者只提供了CellRanger用aggr 方法整合后的文件。也就是说,作者将两个不同组的4个样本的Barcodes,Matrix和Genes文件都整合到了一起,分别提供了三个aggr文件。
在R中用Seruat(5.0.1)读入10X数据
#加载包
library(Seurat)
library(tidyverse)
library(Matrix)
#设置工作目录
setwd("D:/Works/Daily work/Jan_23")
##创建seurat对象
##单细胞数据读取
## read10x
scRNA.counts <- Read10X(data.dir = "D:/Works/Daily work/Jan_23/Mouse")
scRNA=scRNA.counts<-as.data.frame(scRNA.counts)
得到了所有样本的单细胞表达矩阵

#小插曲:在读入数据时,我的输入文件是从GEO下载的上面红框圈起来的3个CellRanger下机文件,R报错:
#但是明明Barcodes文件是存在的,搜索了之后,发现是因为Read10X函数期望读入的是通过早期版本的10X Genomics Cell Ranger软件(<3.0)处理的scRNA序列的文件名(barcodes.tsv,genes.tsv,matrix.mtx),将.gz文件解压并将features.tsv重名为genes.tsv之后读入成功了。
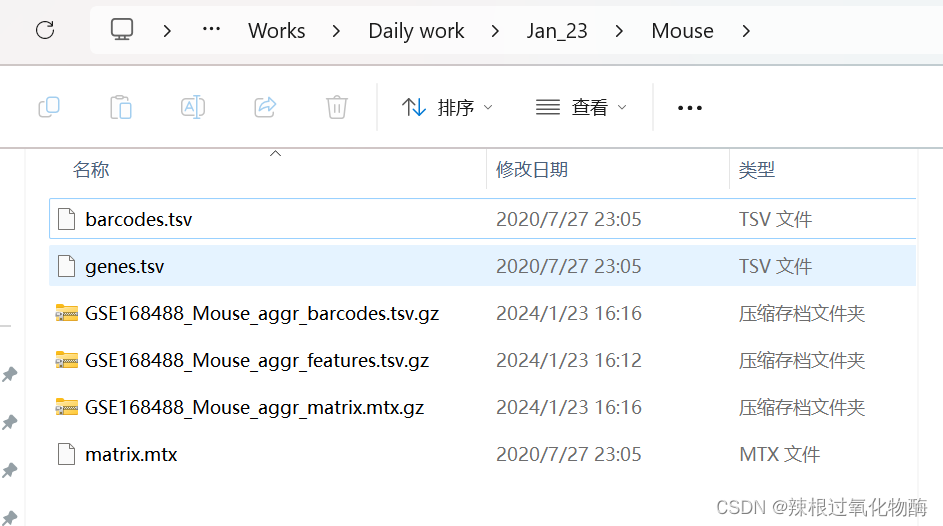
由于接下来要组间的做差异分析(Treated vs Untreated),而我们得到的表达矩阵是来自全部样本细胞的,需要区分样本。上网查阅了很多资料,包括Cellranger的使用手册,并未找到直接拆分aggr文件的方法。
观察Barcodes文件发现,每个细胞Barcodes后的数字为1~4,它们就代表了不同的样本来源。


于是下一步根据细胞barcodes后面的序号拆分样本,之后将4个样本合并到一个Seruat对象scRNAList中,并制作样本的分组信息datMeta文件(这一步是为了方便之后的差异分析)
#前两个是untreated,后两个是treated
U1=scRNA.counts[,grep("-1",colnames(scRNA.counts))]
U2=scRNA.counts[,grep("-2",colnames(scRNA.counts))]
T1=scRNA.counts[,grep("-3",colnames(scRNA.counts))]
T2=scRNA.counts[,grep("-4",colnames(scRNA.counts))]
#sample=c("U1","U2","T1","T2")
#datMeta=as.data.frame(matrix(data=NA,nrow=4,ncol=2))
#colnames(datMeta)=c("sample","group")
#datMeta$sample=c("U1","U2","T1","T2")
#datMeta$group=c(rep("Untreated",2),rep("Treated",2))
#合并样本
scRNAList = list()
scRNAList[[1]] <- CreateSeuratObject(counts = U1,
project = "U1",
min.cells = 3, min.features = 200)
scRNAList[[2]] <- CreateSeuratObject(counts = U2,
project = "U2",
min.cells = 3, min.features = 200)
scRNAList[[3]] <- CreateSeuratObject(counts = T1,
project = "T1",
min.cells = 3, min.features = 200)
scRNAList[[4]] <- CreateSeuratObject(counts = T2,
project = "T2",
min.cells = 3, min.features = 200)可以看到已经成功拆分样本
















 370
370











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









