









单细胞cellranger处理流程
单细胞cellranger处理代码
一套流程直接跑通!
首先你要cd切换到工作目录,需要更改你的reference位置以及制作aggr.csv,然后把下面这些代码复制到一个sc.sh文件中运行就可以了。(没提到的地方都不用改啦)
mkdir SRR
mkdir fastq
#建立两个文件夹
#下载数据
prefetch -O SRR/ --option-file SRR_Acc_List.txt
#SRR_Acc_List.txt大家都会下载吧,在GEO那个数据库,下载后的SRR文件在 SRR/这个文件夹下
#解压sra文件
for i in SRR/SRR*/*.sra; do echo $i ; fastq-dump -O fastq/ --split-files $i; done
#解压的部分用到了fastq-dump,注意后面的参数--split-files,而不是RNA-seq中我们经常使用的--split-3,因为单细胞文件特殊,还含有barcode以及umi文件。
#重命名 适应软件命名需求
cp SRR_Acc_List.txt fastq/
cd fastq
cat SRR_Acc_List.txt | while read i ;do (mv ${i}_1*.fastq ${i}_S1_L001_I1_001.fastq;mv ${i}_2*.fastq ${i}_S1_L001_R1_001.fastq;mv ${i}_3*.fastq ${i}_S1_L001_R2_001.fastq);done
cd ..
#cellranger定量
cat SRR_Acc_List.txt | while read i
do
cellranger count --id=patient$i \
--transcriptome=/PATH/refdata-cellranger-GRCh38-3.0.0 \
--fastqs=fastq \
--sample=$i \
--nosecondary \
--jobmode=local
done
#--nosecondary指的是只获得表达矩阵不进行后续的降维、聚类以及可视化分析
#--jobmode 使用90%的可用内存以及全部核心
#这个部分--transcriptome=/PATH/refdata-cellranger-GRCh38-3.0.0 \你需要给出reference正确的路径
#--id只是一个命名,你可以随意设置只要能够区分即可,会单独形成一个文件夹,详细可见aggr.csv的制作。
#aggregation 整合
cellranger aggr --id=matrix \
--csv=aggr.csv \
--normalize=mapped \
--nosecondary \
#--id指定你输出的文件夹名字,我这里写的是matrix
#--csv 输入的是一个索引,需要你自己制作
#--nosecondary指的是只获得表达矩阵不进行后续的降维、聚类以及可视化分析
aggr.csv的制作
在使用上方代码cellranger aggr的时候需要制作一个类似于索引的csv文件。
我们可以看下cellranger count的输出结果

每一个样本都被单独存放在一个文件夹中,我们进入patient2586outSRR7722937
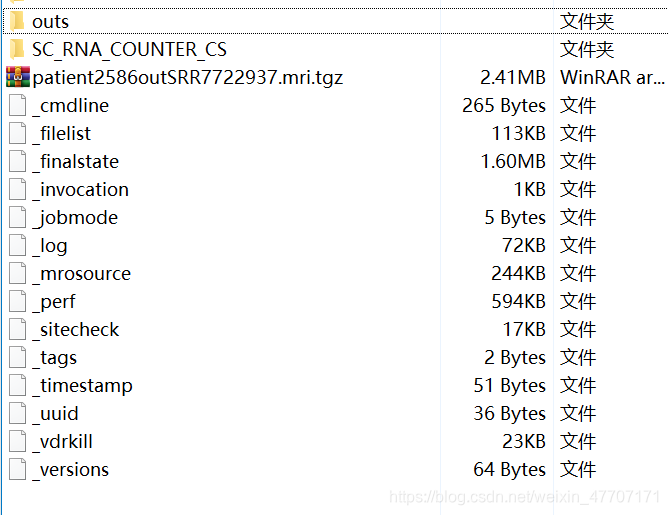
然后我们进入outs文件夹,看到了一个h5文件

那么对应的aggr.csv就应该是这样的,写出样本名以及相对路径即可。
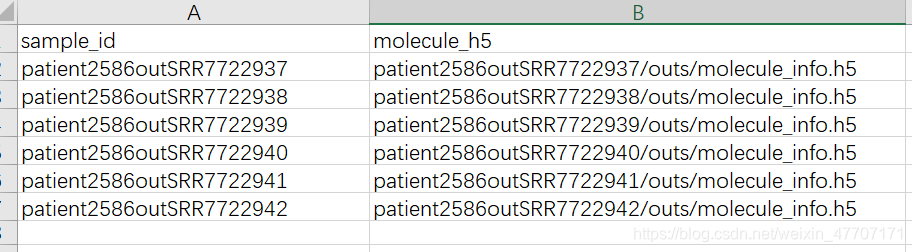
这样,aggr.csv制作完成,运行上方sh文件即可。

















 3391
3391

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









