






 文章讲述了在进行基因组SNPs检测后,尝试使用conda安装的samtools的tview查看比对情况,发现无法实现交互式功能。作者随后选择源码编译安装samtools,但在过程中遇到权限错误,原因是未创建和指定正确的安装路径。经过修正,最终成功安装并能使用交互式的tview功能。
文章讲述了在进行基因组SNPs检测后,尝试使用conda安装的samtools的tview查看比对情况,发现无法实现交互式功能。作者随后选择源码编译安装samtools,但在过程中遇到权限错误,原因是未创建和指定正确的安装路径。经过修正,最终成功安装并能使用交互式的tview功能。
最近在进行基因组SNPs的检测工作,在进行完一个read group的检测工作后,为了了解操作是否正确,想使用samtools tview查看去重复后的比对情况。因为之前吃过环境污染的亏,所以习惯性的使用conda工具进行软件的安装和对应软件工作环境的管理,但是部分软件包由于其特殊性,conda环境下可能某些功能无法实现,比如samtools……
1 conda版的samtools
最初我根据软件说明直接在samtools的环境下查看比对情况:
conda activate samtools
samtools tview SRR098401.dup.bam reference/human_g1k_v37.fasta
发现结果仅是短短一行的非交互式的!!!
网上找了一圈得到的结果是:
通过conda安装的samtools无法实现tview的交互式功能
2 安装samtools时犯病
找到samtools的官网:
下载源码压缩包:
wget https://github.com/samtools/samtools/releases/download/1.17/samtools-1.17.tar.bz2
解压:
tar -jxvf samtools-1.17.tar.bz2
配置环境:
cd samtools-1.17
./configure --prefix=/where/to/install##记住此路径,它是万恶之源!!!!!!!!
编译,安装……
make
make install
就在我傻乎乎的照搬官网的manual的时候,
错误发生了
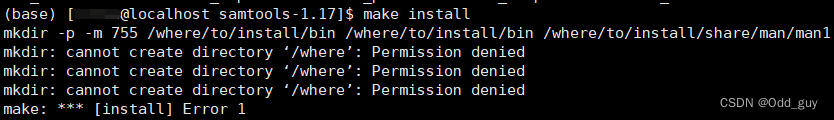
一开始看见Permission denied字样,下意识的以为自己账号权限不够,
至此,已经不再想看比对的情况了,conda版也挺好的,哈哈哈哈……
就在我百无聊赖的看着台历发呆时,无意间瞥见了终端上
报错的信息:
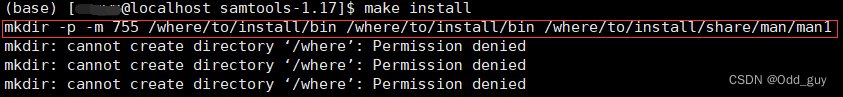
原来是自己在进行环境配置时没有创建和指定安装路径。
3 源码编译正确的打开方式
清楚错误发生的原因后,重新开始安装samtools。
3.1 创建安装目录
mkdir -p ~/samtools/bin
3.2 配置环境
全局变量

重新激活全局变量:
source ./.bashrc
配置编译安装环境
#此路径需要绝对路径,对应安装路径
./configure --prefix=/{your_dir}/samtools
3.3 编译安装
make
make install
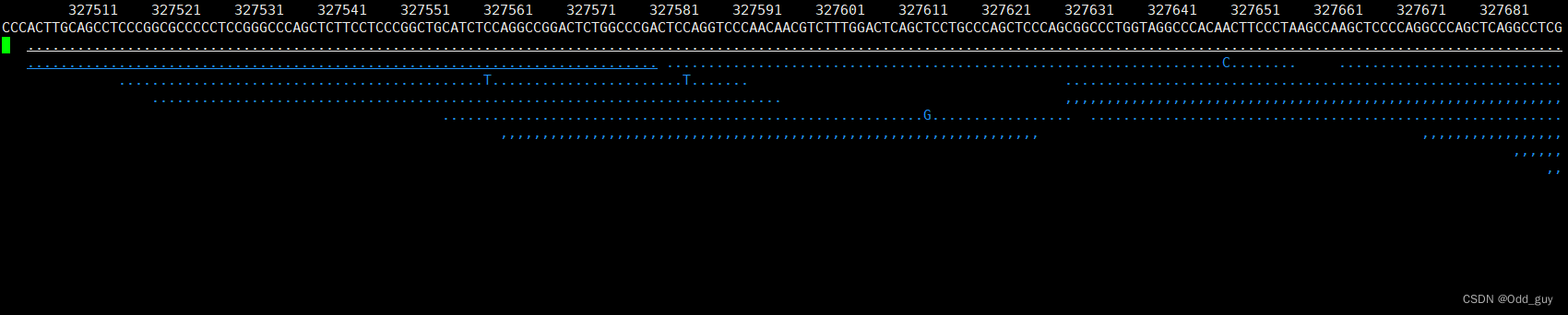
再次运行tview命令
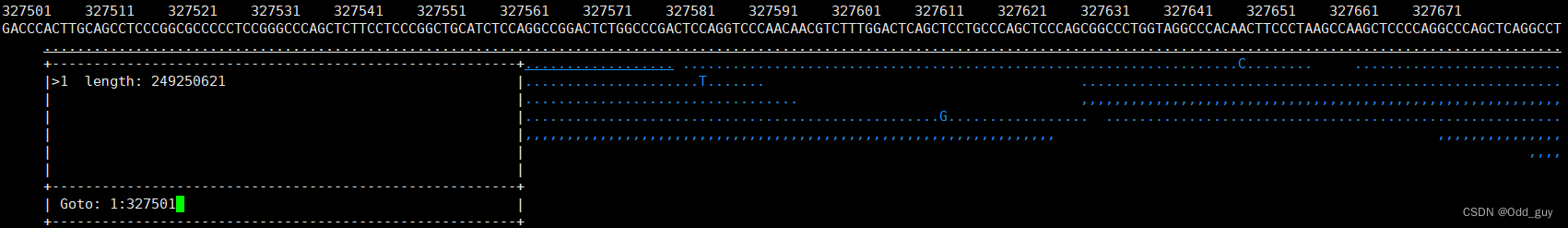
samtools tview SRR098401.dup.bam reference/human_g1k_v37.fasta
可看到交互式的比对内容:




















 2089
2089

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











