







请结合图片,详细解释图片中的内容,要求逻辑清晰,并给出整理与答疑

1,3D结构数据库:



2,PDB文件格式:








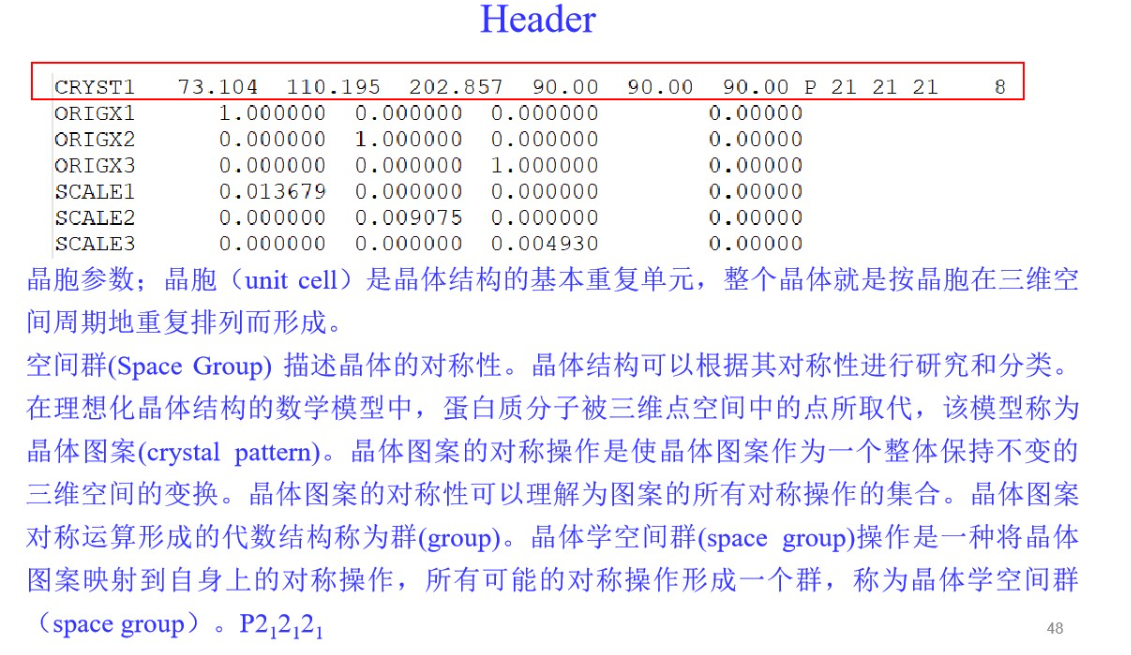


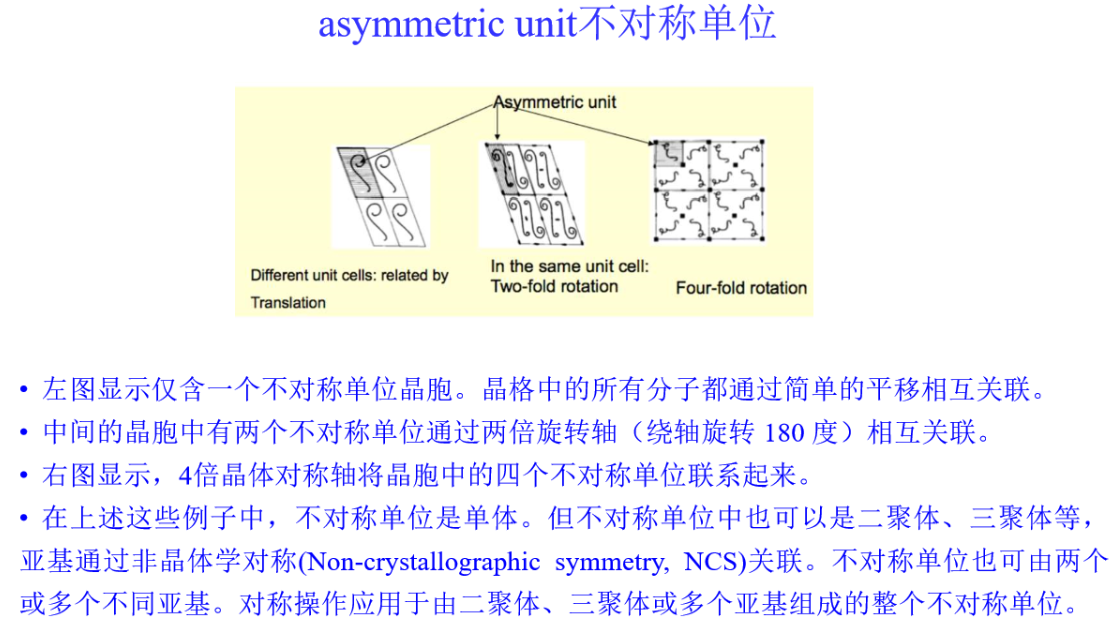


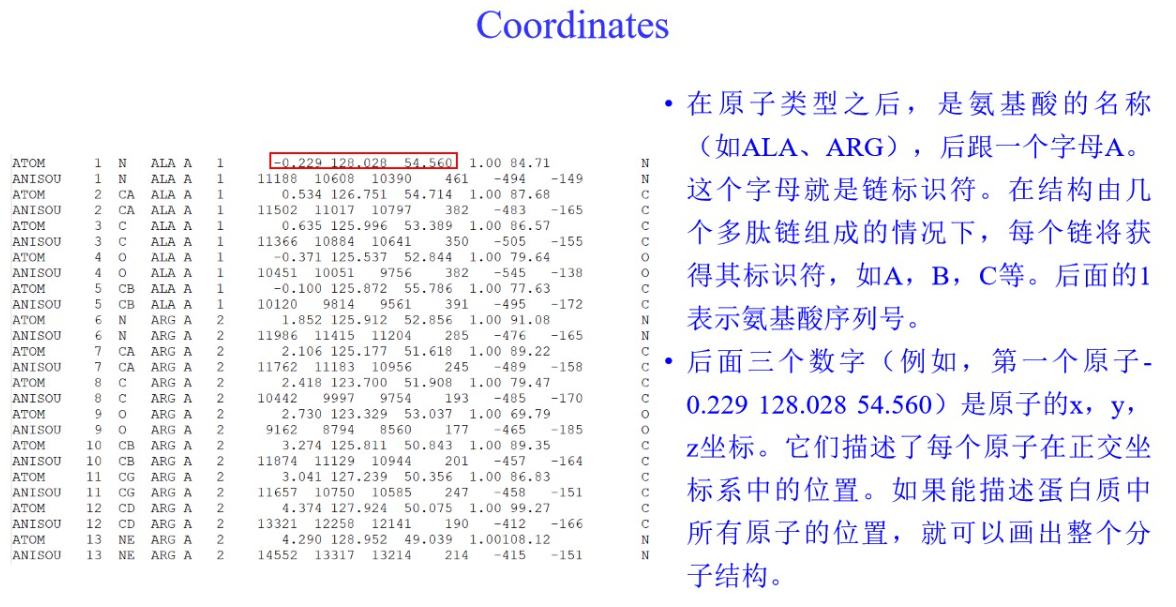

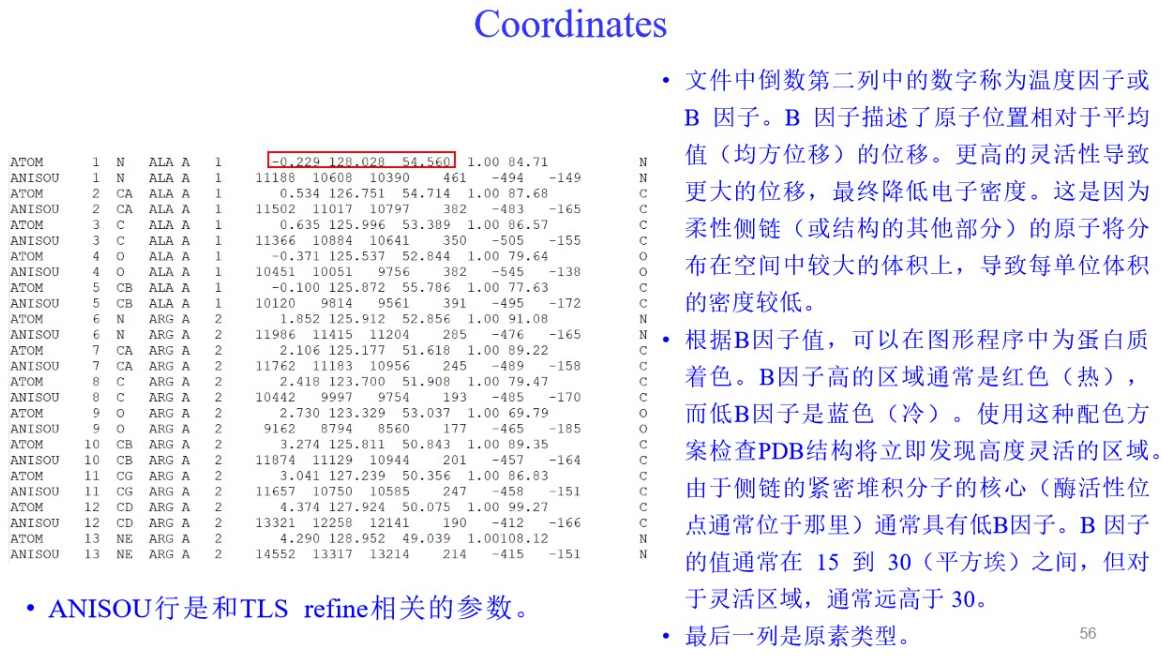





3,PDBX/mmCIF格式:




4,结构因子与密度图:




5,电子云密度图:




(1)2Fo-Fc map、Fo-Fc map



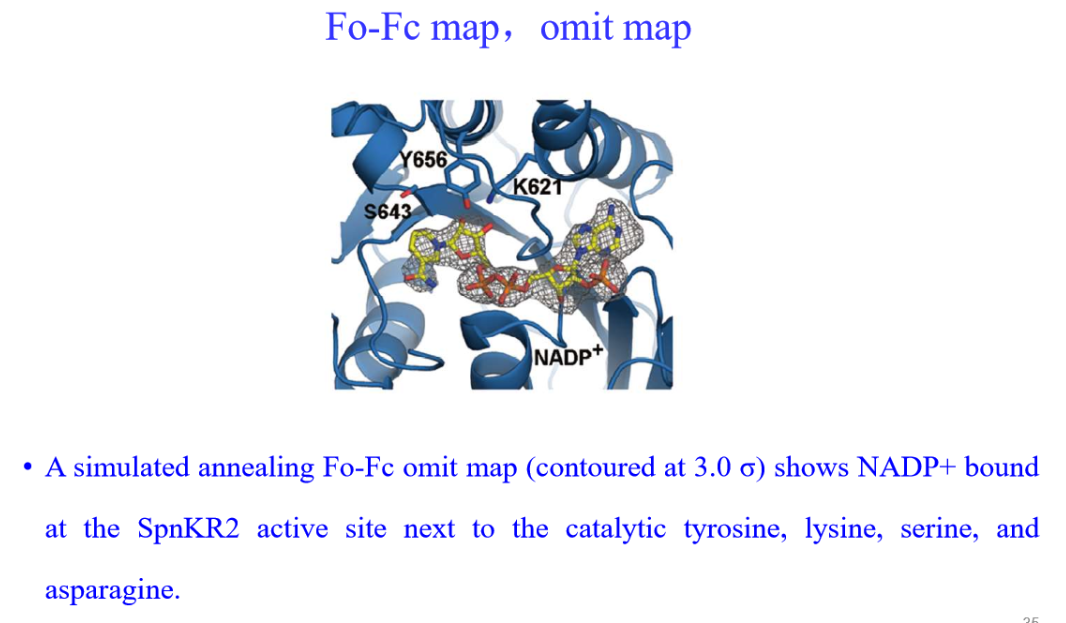


(2)其他:






6,利用coot观察分析模型和密度图:并思考

分析蛋白质结构模型的质量通常涉及以下几个步骤:
- 获取数据:从蛋白质结构数据库(如PDB或mmCIF)获取蛋白质的坐标文件(PDB格式或mmCIF格式)。这些文件包含了蛋白质原子的三维坐标信息。
- 检查坐标文件:通过软件(如PyMol)加载坐标文件,检查模型的立体化学是否合理,包括键长、键角、二面角等。同时,确认没有异常的原子间距离或角度,这些可能指示模型构建错误。
- 获取密度文件:下载相应的电子密度图文件(如MTZ或map格式)。这些文件包含了蛋白质电子密度的分布信息,是验证模型与实验数据一致性的关键。
- 计算结构因子:使用软件(如PHENIX)计算模型的结构因子(Fcalc),并与实验得到的结构因子(Fobs)进行比较。
- 评估模型质量:通过比较Fobs和Fcalc的一致性来评估模型的质量。常用的评估指标包括R因子(R-factor)和相关系数(Correlation coefficient, CC)。
- 相位问题:如果存在相位信息(如从MTZ文件中获得),可以通过相位角的准确性进一步验证模型的质量。
- 模型优化:根据电子密度图和结构因子的比较结果,对模型进行必要的调整和优化。
- 使用图形程序:如Coot或PHENIX,这些程序可以自动执行傅里叶变换,以获得实空间密度,并帮助识别模型与电子密度图之间的差异。
- 分析差异图:Fo-Fc差异图可以揭示模型与实验数据不一致的地方。理想情况下,如果模型与实验数据完美拟合,差异图应该没有显著的密度。
- 使用2Fo-Fc图:2Fo-Fc图可以显示模型与观测数据的拟合程度。在模型构建的中间阶段,这个图可以帮助识别模型中可能缺少的特征。
- 检查电子密度图:通过检查2Fo-Fc和Fo-Fc电子密度图,可以发现模型无法解释的密度区域,或者模型与电子密度不匹配的区域。
- 最终验证:在模型优化结束后,应该再次检查2Fo-Fc图,确保没有明显的没有相应模型的密度,这表明模型与实验数据有很好的一致性。
===================================================================
7,可视化工具:pymol主


phenix:上游

CCP4:更像是个套件(上游汇总)

coot:上游结构解析
pymol:解析结构的下游,其他都是上游工具(下游可视化)

==================================================================























 1353
1353

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









