







背景

无论是在进行细菌、古菌高通量测序前,还是细菌、古菌qPCR前,最纠结的问题永远是引物选择。评估引物效果最重要的两个指标是覆盖率(coverage)和特异性(specificity)。简单讲,覆盖率就是指目标引物能捕获现有数据库中靶序列的比例,比如,共有100种细菌的不同16S rRNA基因序列,某引物能扩增出其中的90种,那么该引物的覆盖率就是90%;特异性是指目标引物是否只靶标某特定类群,比如,某古菌引物PCR获得的序列全部为古菌,没有任何细菌或其他非古菌序列,则其特异性为100%。本文将介绍一款超级简单、快速、准确地评估16S rRNA基因引物覆盖率及特异性的工具。
TestPrime
该工具是SILVA官网发布的一款在线工具,名为TestPrime 1.0,其网址为:https://www.arb-silva.de/search/testprime/,打开网址后其界面如下图1。由图可知,主要包括四部分:①Sequence Data,需要将待检测的Forward Primer及Reverse Primer序列分别按照5'--3'的方向在此输入;②Database,在此选择合适的数据库及比对序列,一般默认为SILVA最新释放版本(目前为r132)的非冗余参考序列(RefNR);③Mismatches,可设置所允许的引物最大错配数及3'端0错配的长度;④Job name,可给当前任务命名。
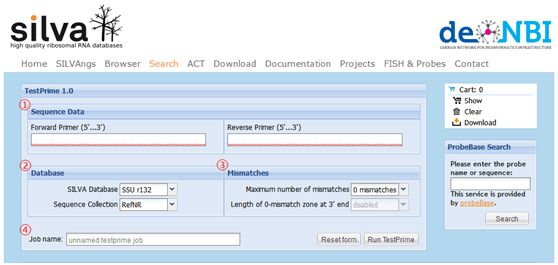
图1. TestPrime 1.0的主界面
使用
将待检测的Forward Primer及Reverse Primer序列分别按照5'--3'的方向输入上图“Sequence Data”部分对应文本框中→在“Database”处选择合适的数据库及比对序列→在“Mismatches”处,设置所能允许的引物错配值→在“Job name”处填写此次任务名→点击界面右下角“Run TestPrime”→静待片刻(依网速几分钟到十几分钟),收获结果。
运行结束后,可在页面下方的“TestPrime Tasks”看到类似图2所示界面,点击红色箭头处“show result”,即可跳转至相应任务的结果页面。
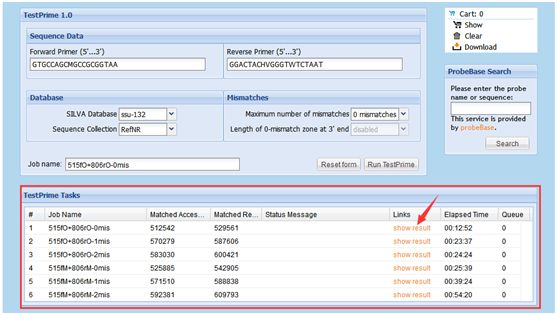 图2. TestPrime运行任务列表及进程示例
图2. TestPrime运行任务列表及进程示例
下拉页面即可看到不同的结果展示内容,主要包括“Overview (all domains)”、“Table of Matched Accessions per Taxonomy”、“Table of Matched Regions”三部分内容(图3)。其中“Table of Matched Accessions per Taxonomy”部分(图3红框标出)就包含引物与SILVA数据库比对后的各分类级别(domain到genus)的覆盖率及特异性等信息。点击左下角“Export Table to CSV”即可导出CSV格式结果数据,用Excel打开文件即可。
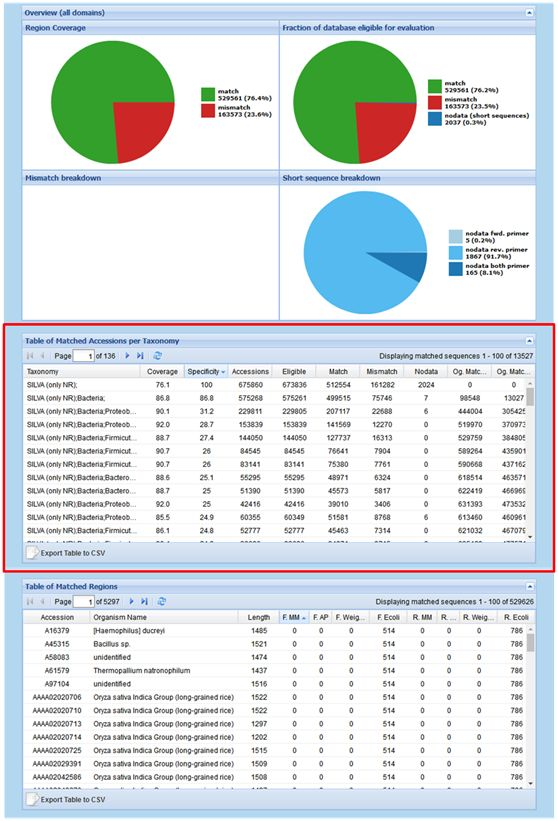
图3. TestPrime运行结果示例
示例
现以Walters et al. (2016)文章提到的两对常用细菌&古菌通用引物为例(表1),比较新旧引物的覆盖率变化。Walters等在其文章中提到,相比最初的引物515f Original+806r Original,各改变一个碱基后的新引物515f Modified+806r Modified会显著提高对细菌SAR11 clade和古菌Thaumarchaeota的覆盖率。这个结论能否通过TestPrime再现呢?
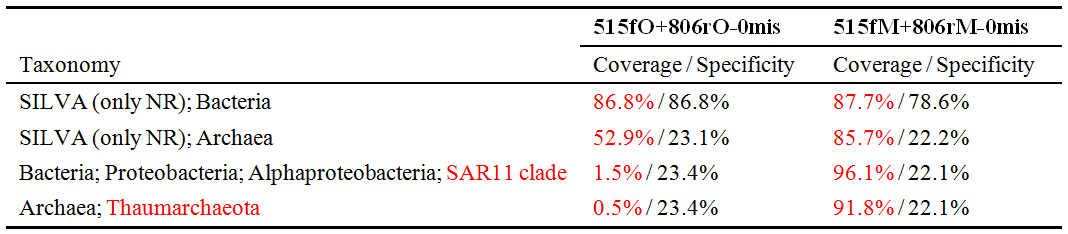
我们分别将两对新旧引物(见表1)通过TestPrime检测其覆盖率,从输出结果中分别将两对引物对总细菌、古菌及所关注的细菌SAR11 clade和古菌Thaumarchaeota的覆盖率整理为表2,可发现:新引物对(515fO+806rO)相比旧引物对(515fM+806rM)在0错配(0mis)情况下,虽然对总Bacteria的覆盖率仅由86.8%提高至87.7%,但对细菌SAR11 clade的覆盖率从原来的1.5%显著提高至96.1%!对总Archaea的覆盖率从原来的52.9%显著提高至85.7%,尤其是对古菌Thaumarchaeota从原来0.5%提高至91.8%!因此,通过TestPrime依据SILVA数据库的引物检测结果与Walters等在其文章中提到的结果一致!
表1. 细菌&古菌通用引物列表
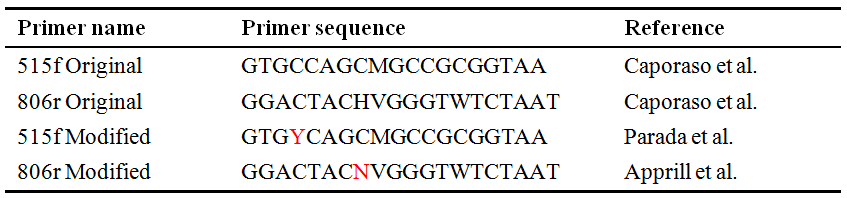
表2. TestPrime检测细菌&古菌新、旧通用引物结果中摘录的部分类群覆盖率
更多
1. 图1“Database”处,可选择SSU(16S / 18S)或LSU(23S / 28S),因此,TestPrime除了量化检测16S rRNA基因引物的覆盖率、特异性等,还可以检测18S、23S或28S相关的引物对。
2. 对于SSU / LSU相关的探针或单引物的覆盖度及特异性检测,可用SILVA开发的另一款在线工具TestProbe 3.0(https://www.arb-silva.de/search/testprobe/probe),用法类似,不再赘述。
3. 更详细的教程及更多的引物评估相关参数请参见官网说明:https://www.arb-silva.de/documentation/testprime-tutorial/
4. 引物真实效果与多种因素相关,通过数据库来看覆盖率和特异性只是其中重要一方面,真实效果要合成引物后结合实际实验去验证。
参考文献
Apprill A, McNally S, Parsons R, et al. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton[J]. Aquatic Microbial Ecology, 2015, 75:129-137.
Caporaso J G, Lauber C L, Walters W A, et al. Global patterns of 16S rRNA diversity at adepth of millions of sequences per sample[J]. Proceedings of the National Academy of Sciences, 2011, 108(Supplement 1): 4516-4522.
Klindworth A, Pruesse E, Schweer T, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies[J].Nucleic Acids Research, 2013, 41(1): e1-e1.
Parada A E, Needham D M, Fuhrman J A. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples[J]. Environmental Microbiology, 2016, 18(5): 1403-1414.
Walters W, Hyde E R, Berg-Lyons D, et al. Improved bacterial 16S rRNA gene (V4 and V4-5)and fungal internal transcribed spacer marker gene primers for microbial community surveys[J]. mSystems, 2016, 1(1): e00009-15.


本期校稿:卢瑟菌 李小圆
本期排版:李小圆
猜你喜欢
10000+:肠道细菌 人体上的生命 宝宝与猫狗 梅毒狂想曲 提DNA发Nature 实验分析谁对结果影响大 Cell微生物专刊
文献阅读 热心肠 SemanticScholar Geenmedical
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外150+ PI,1500+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”



















 123
123

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









