






 本文展示了如何使用R包ggtern和amplicon复现微生物相对丰度的三元相图,通过处理数据、计算相对丰度和选择高丰度门,最终绘制出信息丰富的三元图。
本文展示了如何使用R包ggtern和amplicon复现微生物相对丰度的三元相图,通过处理数据、计算相对丰度和选择高丰度门,最终绘制出信息丰富的三元图。
本文代码已经上传至https://github.com/iMetaScience/iMetaPlot230125ternary 如果你使用本代码,请引用:Chenyuan Dang. 2022. Microorganisms as bio‐filters to mitigate greenhouse gas emissions from high‐altitude permafrost revealed by nanopore‐based metagenomics. iMeta 1: e24 https://doi.org/10.1002/imt2.24
代码编写及注释:农心生信工作室
写在前面
三元相图 (Ternary plot) 可以展示三个变量的关系,既美观且信息更加丰富,在微生物领域常用于比较多个样品的丰度信息。本期我们挑选刊登在iMeta上的Microorganisms as bio‐filters to mitigate greenhouse gas emissions from high‐altitude permafrost revealed by nanopore‐based metagenomics.论文的Figure 2A进行复现,基于ggtern绘制三元图,先上原图:

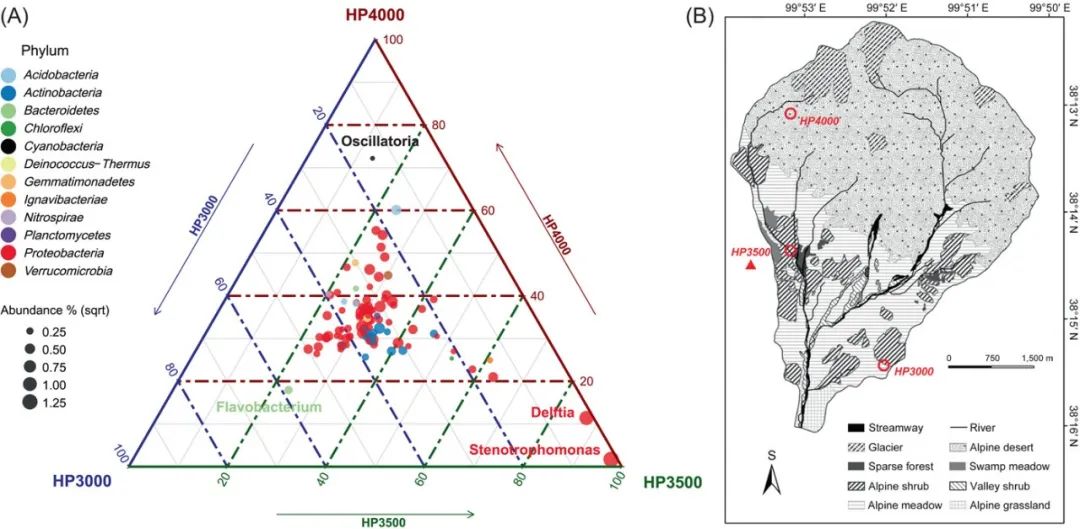
接下来,我们将通过详尽的代码逐步拆解原图,最终实现对原图的复现。
R包检测和安装
01
安装核心R包ggtern以及一些功能辅助性R包,并载入所有R包
options(repos = list(CRAN = "https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
if (!require("devtools"))
install.packages('devtools')
if (!require("ggtern"))
install.packages('ggtern')
install.packages("devtools")
devtools::install_github("microbiota/amplicon")
# 加载包
library(amplicon)
library(ggtern)读取数据及数据处理
02
使用R包amplicon中的示例数据tax_phylum并计算相对丰度
dim(tax_phylum) # 查看数据维度,共3个样品各6个重复共18列数据
#> [1] 18 19
rela_tax <- apply(tax_phylum, 2, function(x) x/sum(x)) # 绝对丰度/对每一列的丰度总和得到每个物种的相对丰度
rela_group_tax <- t(apply(rela_tax, 1, function(x) c(mean(x[1:6]), mean(x[7:12]), mean(x[13:18])))) # 对每个样品的6个重复计算相对丰度均值
colnames(rela_group_tax) <- c('KO', "OE", "WT") # 对列重命名03
保留相对丰度较高的10个门,合并其余门的相对丰度
df <- as.data.frame(rela_group_tax)
df$total <- rowSums(df) # 对每个门额外计算一列总相对丰度用于选出高丰度的门
sort_df <- df[order(df$total, decreasing = T), ] # 得到排序后的数据框
others <- colSums(sort_df[11:nrow(df), ]) # 计算其他低丰度门的丰度总和
df2 <- rbind(sort_df[1:10, ], others) # 合并表格
# 计算丰度并添加分组,分别代表三元图中点的大小和颜色
df2$Abundance <- rowMeans(df2)
gp <- c(rownames(df2)[1:10], "Others")
df2$Phylum <- factor(gp, level = gp)绘图预览
04
使用ggtern包绘制群落丰度三元图:
p <- ggtern(data = df2, aes(KO, OE, WT)) + geom_point(aes(color = Phylum, size = Abundance)) + theme_bw() + theme_arrowdefault()
ggsave('ternay.png', p, width = 10, height = 8)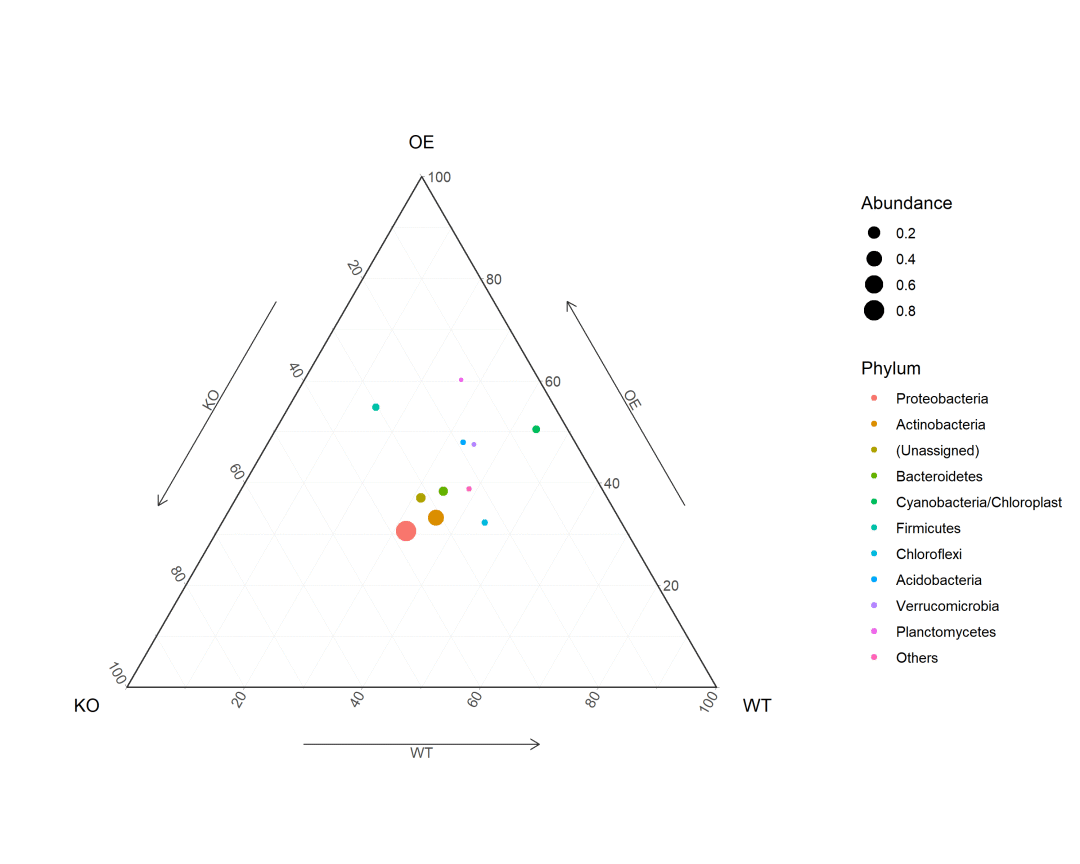
完整代码
options(repos = list(CRAN = "https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
if (!require("devtools"))
install.packages('devtools')
if (!require("ggtern"))
install.packages('ggtern')
install.packages("devtools")
devtools::install_github("microbiota/amplicon")
# 加载包
library(amplicon)
library(ggtern)
dim(tax_phylum) # 查看数据维度,共3个样品各6个重复共18列数据
#> [1] 18 19
rela_tax <- apply(tax_phylum, 2, function(x) x/sum(x)) # 绝对丰度/对每一列的丰度总和得到每个物种的相对丰度
rela_group_tax <- t(apply(rela_tax, 1, function(x) c(mean(x[1:6]), mean(x[7:12]), mean(x[13:18])))) # 对每个样品的6个重复计算相对丰度均值
colnames(rela_group_tax) <- c('KO', "OE", "WT") # 对列重命名
df <- as.data.frame(rela_group_tax)
df$total <- rowSums(df) # 对每个门额外计算一列总相对丰度用于选出高丰度的门
sort_df <- df[order(df$total, decreasing = T), ] # 得到排序后的数据框
others <- colSums(sort_df[11:nrow(df), ]) # 计算其他低丰度门的丰度总和
df2 <- rbind(sort_df[1:10, ], others) # 合并表格
# 计算丰度并添加分组,分别代表三元图中点的大小和颜色
df2$Abundance <- rowMeans(df2)
gp <- c(rownames(df2)[1:10], "Others")
df2$Phylum <- factor(gp, level = gp)
p <- ggtern(data = df2, aes(KO, OE, WT)) + geom_point(aes(color = Phylum, size = Abundance)) + theme_bw() + theme_arrowdefault()
ggsave('ternay.png', p, width = 10, height = 8)以上数据和代码仅供大家参考,如有不完善之处,欢迎大家指正!
更多推荐
(▼ 点击跳转)
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法
iMeta | 浙大倪艳组MetOrigin实现代谢物溯源和肠道微生物组与代谢组整合分析

第1卷第1期

第1卷第2期

第1卷第3期

第1卷第4期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science

















 1400
1400

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









