






识别生物时间序列中的局部关联:算法、统计显著性和应用
Identifying local associations in biological time series:
algorithms, statistical significance, and applications

Article,2023-11-2,Briefings in Bioinformatics, [IF 9.5]
DOI:https://doi.org/10.1093/bib/bbad390
原文链接:https://academic.oup.com/bib/article/24/6/bbad390/7337690
第一作者:艾冬梅
通讯作者:夏立
- 摘要 -
局部关联是指生物领域出现的时空相关性,如时间依赖性基因共表达或微生物之间的季节性相互作用。人们可以通过检查这些关联的生物时间序列数据来揭示生物系统的复杂动态和内在相互作用。为了实现这一目标,已经开发了局部相似性分析算法和统计方法,以促进时间序列的局部对齐并评估结果对齐的意义。虽然这些算法最初是为微阵列的基因表达分析而设计的,但它们已经适应并加速用于多组学下一代测序数据集,产生了很高的科学影响。本文综述了局部相似度分析算法的历史发展和最新进展、它们的统计特性以及在生物时间序列数据分析中的实际应用。本文中使用的基准数据和分析脚本可以在http://github.com/labxscut/lsareview上免费获得。
关键词:时间序列数据;局部关联;局部对齐;局部相似性分析;局部趋势分析;统计显著性
- 引言 -
在微生物组或转录组的时间序列数据中识别生态和生化关系通常通过基于相关性的共发生和共表达分析来实现。然而,全局相关度量(如Pearson或Spearman相关性)只有在两个时间序列全局同步和关联时才有效。然而,在真实的生物数据中,更复杂的动态变化关系普遍存在,包括局部关联和时间延迟关联,这些关联已在微生物生态学[1-4]、分子生物学[5,6]和功能神经科学[7,8]等多个领域被观察到。例如,ARG2(乙酰谷氨酸合成酶)和CAR2(鸟氨酸转氨酶)基因表达分析中的调控机制取决于CPA2(精氨酸特异性氨甲酰磷酸合成酶)的表达水平。尽管如此,ARG2和CAR2之间的全局Pearson相关系数(PCC)表达水平几乎为零[9],这强调了全局相关方法在检测局部关联方面的不敏感性。尽管全局相关性仍然是有价值的工具,但有必要制定更灵活的替代假设来检测生物时间序列中存在的复杂关系。
为了解决全局相关性的局限性,研究人员引入了许多方法,其中最突出的是局部相似性分析(LSA)[10-13]。LSA是一种基于局部对齐的方法,用于识别针对两个时间序列之间的局部相似性分数进行优化的对齐配置,从而能够检测局部和潜在时间延迟的成对关联。一种相关的方法,被称为局部趋势分析 (LTA)[14-16],是LSA的一种变体,它识别趋势序列对之间的对齐配置和相关性,代表上升、下降和不变化状态。LSA和LTA也会评估所识别的关联的统计显著性。值得注意的是,术语局部相似性不仅用于生物学[17],还用于化学[18]、公共卫生[19]和电力工程[20]等领域,只是含义不同。我们建议读者参考他们各自的论文和评论。
本文专门研究生物时间序列中局部相似性的背景。在生物时间序列领域,局部相似性分析在揭示微生物物种丰富度与环境因子之间的潜在关系方面发挥着关键作用。当实际相关性限于特定的时间子区间或相关性受时间延迟影响时,局部相似性变得十分有意义[10]。局部相似性分析在不同环境下微生物群落的时空演化研究中得到了广泛的应用。重要的是,通过传统的基于相关性的分析方法无法有效地识别这些微生物物种之间的潜在关系。
还有其他方法可以识别不同类型的复杂关联[9,21 - 23]。例如,液体关联(LA)技术[9,22,24]检测三元组而不是成对关联,而最大信息系数(MIC)方法识别未指定的一般成对关联[25]。格兰杰因果分析可以识别分子生态时间序列数据中潜在的因果关系[26]。此外,LA与LSA相结合可用于检测检测介导的协变动力学,从而发现第三方中介变量[27]。本文综述了LSA和LTA的研究进展,包括算法、统计显著性评价方法、软件工具的可用性及其应用。文献[28,29]提供了有关相关测度及其应用的进一步信息。
LSA建立在局部序列对齐的概念之上,利用一种类似于SmithWaterman算法[17]的动态规划算法来检测使局部相似性得分最大化的最优时间序列对齐配置。Qian等人首先介绍了基因表达谱分析方法[10]。基于该方法,LSA被扩展到分析微生物组数据,包括Ruan 等人[13]的操作分类单位(OTU)分析和Xia等人[12]的宏基因组数据分析。对LSA的方法研究也取得了进展,包括Xia等人[12]对重复序列数据的支持,Xia等人[11]和Durno等人[30]报道的统计显著性理论近似(即p值),以及更先进的p值计算方法,如Zhang等人提出的移动块抽样LSA (MBBLSA)[31]和数据驱动LSA (DDLSA)[32]。
伴随着许多其他最近开发的用于从时间序列数据中提取有意义的关联模式的相关性测量技术,Ruan等人的LSA[13]和Xia等人的扩展的局部相似性分析工具eLSA[11,12]仍然具有优势。例如,Wang等人[33]开发了一种计数统计,并将其与LSA进行了比较,结果表明,随着相关子序列对的噪声增加,LSA始终保持鲁棒性并变得更有效。同样,Tackmann等人[34]引入了一种Flashweave算法,该算法旨在推断高分辨率交互网络,而在使用Weiss等人模拟的真实数据进行基准测试时,eLSA的性能优于该算法[28]。
自2011年以来,eLSA工具已广泛用于微生物数据分析,导带来了许多发现。如Liu等人[35]利用eLSA分析了水库中细菌OTUs与浮游植物之间的相关性,得到了一个具有显著局部生态关联的动态相互作用网络。同样,Thiriet-Rupert等人[36]构建了微藻的基因共表达网络,通过eLSA确定差异表达的转录因子与突变表型之间的关系。Lee等[37]利用eLSA对柴油污染土壤根茎修复、环境条件和细菌属之间的时间依赖关系进行了评价,得到了相关网络。
此外,eLSA已被用于多种学科的研究,包括Parada等人[38]研究海洋古生物与微生物群落的相关性,Jones等人[39]推断微生物的多样性生态关系,Liang等人[40]分析消化污泥微生物分类群的关联网络,以及识别海洋浮游植物、细菌和病毒之间的复杂关系[41,42]。病毒噬菌体与宿主之间的关系[43],以及抗生素耐药基因与细菌群落之间的关系[44]。
此外,eLSA识别时移相关性的能力也得到了广泛的应用。例如,Posch等[45]利用eLSA构建了纤毛虫与浮游植物之间的相互作用网络,研究了具有时滞的相关性。他们在纤毛虫之间以及纤毛虫和藻类之间发现了这种联系。Džunková等人[46]研究了考虑时间延迟的不同个体口腔细菌OTUs之间的相关性。在另一个例子中,eLSA被用于评估活性污泥微生物成员与环境变量之间的时滞相关性[47]。
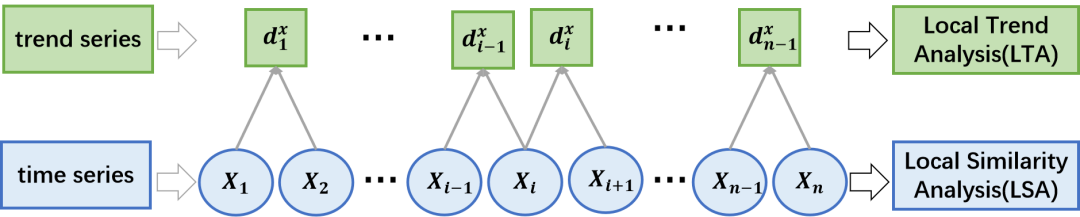
与LSA有关的研究鼓励开发分享其原理。其中一种方法是局部趋势分析(local trend analysis, LTA),由He等[14]和Ji等[15]提出,用于分析基因表达谱序列的变化趋势。在LTA中,对经过趋势变换的原始数据序列采用局部对齐算法。趋势转换包括取相邻时间点的值差并将其离散化成{1,0,−1},分别代表{上升,不变,下降}的趋势状态。Xia等[16]提出了一种近似LTA统计显著性的理论,Shan等进一步完善了该理论,称为平稳理论局部趋势分析(stationary theoretical local trend analysis,STLTA)[48]。
与传统的全局相关测量相比,大量研究已经证明了LSA和LTA在推断时间依赖性关联方面的有效性。增加的时间维度有助于挖掘延迟背后的因果关系。此外,统计显著性的高效理论近似,即p值,在LSA和LTA分析的广泛使用中发挥了关键作用。这些方法已成功应用于基于大规模下一代测序(NGS)的数据集,否则使用更慢的基于排列的p值进行分析是不切实际的。在本文中,我们深入介绍了LSA和LTA的相关算法和理论,以及它们的最新改进,包括eLSA、fastLSA、MBBLSA、DDLSA和STLTA。
- 生物序列数据的局部相似性分析 -
生物时间序列数据
时间序列数据是研究生物系统动力学的重要资源。这些数据可能是基因调控研究中的基因表达水平,也可能是生态学研究中的环境和生物因子水平。时间序列数据的性质可能是高度可变的,根据数据集的不同,其值表示不同的测量值。
在对原始数据集进行LSA处理之前,需要对数据进行归一化处理。此步骤将原始值转换为平均值为零,方差为1的标准化值。然而,标准化基于计数的时间序列数据的一个障碍是零的高频率。从归一化过程中排除零值可能是优先的,因为这些值可能是由于有限的检测灵敏度或随机丢失而不是真正的不存在。可以使用各种归一化方案,例如z分数归一化和百分位数归一化。Xia等人使用了带有稀疏度调整的百分位数归一化方法,这种方法通常在数据集之间提供最佳和最稳健的结果,即使对于具有非线性关联的数据集也是如此(详细步骤参见论文[12])。
生物序列数据的局部比对和相似性得分


Note: sgn(.) is the sign function.
与Smith-Waterman算法相比,该局部对齐算法具有独特的特点。首先,引入最大时间延迟D,将动态规划结果限制在起始点X和起始点Y相距最大D个时间单位的对齐上。D通常很小,因为延迟关联通常表示对最近其他因素变化的即时反应,滞后时间通常很短。一个小的D也有效地减少了算法的搜索空间,对于输入数据的大小,算法的计算复杂度和空间复杂度从的二次型降低到线性型。其次,除了优化一个目标,还有两个得分矩阵,P和N,同时更新以实现最大化:一个用于正向关联,另一个用于负向关联。局部相似度S,计算为P和N中所有条目的绝对值的最大值。S通过对级数√n取平方根来标准化。
局部相似度评分最初按n进行缩放,参考[11-13]。然而,回顾过去,当比较不同系列长度的分数时,√n更可取[26]。今后,我们将把这个√n倍的对齐分数称为局部相似性分数或LS分数。值得注意的是,与Pearson’s或Spearman’s相关性不同的是,这个LS分数并没有限定在- 1到1的范围内。
重复序列数据的局部相似性评分
通过重复评估可变性对于统计推断非常重要[49-51]。原来的LSA方法只考虑序列数据,不考虑重复。Xia等人首先提出了扩展局部相似性分析(extended local similarity analysis, eLSA)方法,将具有重复的序列数据纳入其中[12]。eLSA假设每个样本有m个重复,并使用函数 F (·)来总结重复的测量。算法2提出了一种改进的eLSA动态规划算法。
在改进后的算法中,将原始LSA算法中的标量Xi 和 Yi替换为总结和变换后的值F(Xi ) 和 F(Yj ),分别应用于大小为1×m的m个复制数据向量Xi和 Yj。因此,当m = 1时,eLSA框架将原始LSA视为其特例。

Note: sgn(.) is the sign function.
转换函数用于重复数据
为了对eLSA的重复测量和可变性进行建模,Xia等人引入了汇总函数F(·)。研究者此前提出了几种方法[51-53]来完成这项任务,包括简单均值法(Mean)、标准差加权均值法(SD)[52]以及与“均值”法相同的多变量相关系数法[54]。此外,与传统方法相比,稳健统计不需要正态性假设。它们对数据中异常值的影响表现出有效的弹性,更准确地反映了实际情况[54]。例如,在使用样本时对于总体均值估计,异常值的存在显著影响估计结果。相反,样本中位数对数据异常值的敏感性较低,使其比样本平均值具有更强的鲁棒性[55]。因此,稳健统计,如中位数(Med)和中位数绝对偏差(MAD)是“Mean”和“SD”的稳健替代[56],得到的汇总函数被纳入eLSA,分别称为中位数法(Med)和MAD加权中位数法(MAD)。
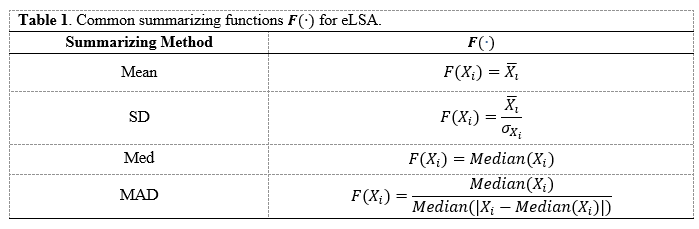
局部相似度评分的Bootstrap置信区间

局部相似度评分显著性的统计理论
置换统计显著性
非复制数据
在原始非复制数据LSA分析中,LS评分的统计显著性(p值)是通过置换计算的。该过程包括以下步骤:
(i)对LS分数为S(X,Y)的每对时间序列X和Y,对每个序列中的数据值进行排列,生成一个置换的样本对。
(ii)使用LSA算法(算法1)计算排列后样本的LS评分。
(iii)重复步骤(i)和(ii) L次,得到(X1,X2,…,XL ) 和 (Y1,Y2…YL),表示X、Y的L个置换样本,并计算其排列后的LS分数。
(iv) p值确定为至少与S (X, Y)相同的排列LS分数的比例,即:
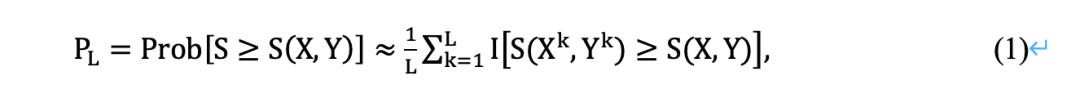
其中I(·)为示性函数。
复制数据
在复制数据eLSA分析的情况下,对置换检验进行修改,使每个排列样本的序列数据Y保持不变,而对X的所有列进行重新排列。对于固定数量的排列L, (X1,X2…XL )是X的置换样本,p值PL𝐿的计算方法为:
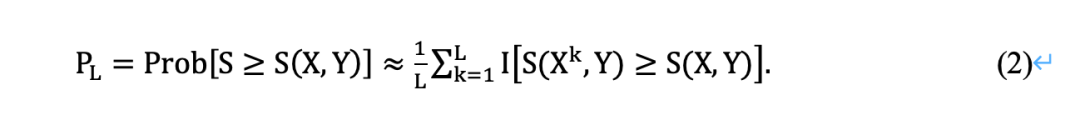
滑动分块自助法


置换和bootstrap方法的瓶颈

局部相似性分数的理论统计显著性
Feller关于随机游走偏移范围的理论
由于计算复杂性,基于置换的步骤仅适用于具有几百个因素的成对LSA分析。因此,有必要发展有效的理论p值近似来缓解这一问题。一个方向是将Feller关于随机游走范围的理论应用到LSA的设置中。Feller[58]研究了n个期望为零的随机变量的和的范围的近似分布。设Z_i为独立同分布 (i.i.d.)随机变量,使得E(Zi )=0 , Var(Zi )=σ2,其中E(∙)是数学期望, Var(∙)是方差。设Cn=Z1+Z2+⋯+Zn,Mn=max{0,C1,C2,⋯,Cn}, mn=min{0,C1,C2,⋯,Cn }。偏移范围定义为Rn=Mn-mn。根据Feller[56],可以得出:
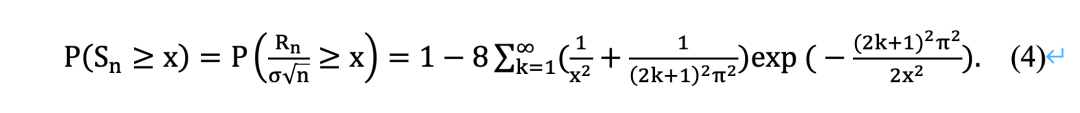
一种局部相似度评分理论

局部相似度评分理论的推广
Xia等[11]提出的方法仅对独立且同分布(i.i.d)序列有效。然而,在实践中,许多序列并不是独立同分布。为了解决这一问题,Zhang等[32]引入长期方差ω2,用 ω 代替 σ,利用Eq.(4)近似p值。
准确估计ω对于评估相关序列的LS评分的统计显著性至关重要。Zhang等人[32]应用了数据依赖的AR(1)插件[59],得到的长期方差估计为:
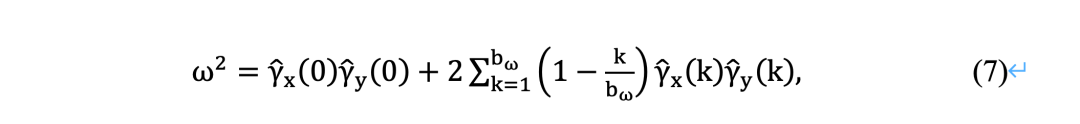

统计显著性的上界方法


- 统计显著性近似方法的比较 -
我们比较了五种可用的开源LSA软件工具,它们实现了估计LS分数的统计显著性的置换方法或理论方法,如表2所示。这些工具包括eLSA[11,12]、fastLSA[30]、LocSim[13]、MBBLSA[31]和DDLSA[32]。eLSA和fastLSA都是用高效的c++代码实现的,而LocSim、MBBLSA和DDLSA都是用r语言编写的。eLSA提供了理论计算(Xia等人的[11])和基于置换的p值计算。FastLSA和DDLSA提供了理论上的p值计算。而LocSim和MBBLSA只实现了排列方法。eLSA还包括其他特性,如趋势序列分析、复制数据分析和 bootstrap置信区间。此外,我们提供了图1作为一个可行的决策树,以便为各种任务或数据集选择合适的软件工具。

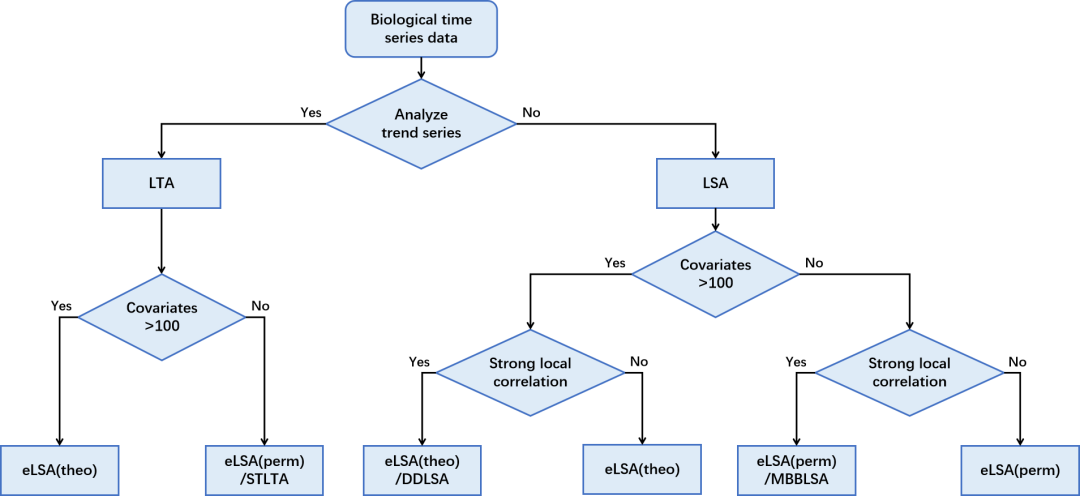
图1.选择局部相似性分析软件工具的决策树。
此外,我们比较了p值近似方法的性能:理论或置换。理论方法分别在eLSA (Xia等人[11])、fastLSA (Durno等人[30])和DDLSA (Zhang等人[32])中实现。置换方法在LocSim(Ruan等人[13])和MBBLSA(Zhang等人[31])中实现。LocSim R工具被排除在这个比较之外,因为它的排列方法与eLSA相同,但速度较慢。
我们从GitHub (https://github.com/wdwvt1/correlations)[28]获得了Weiss等人准备的基准数据。出于本文的基准测试目的,我们使用了他们的一部分数据,如表3所示。表S1-4(原Weiss et al.的表2.6-2.9)模拟了不同约束条件下两个物种的Lotka-Volterra关系。Lotka-Volterra关系通过模拟生态关系的n个微分方程系统描述了n个物种的丰度。这些表中模拟的正Lotka-Volterra关系与来自对数正态分布和伽马分布的模拟随机协变量(即OTU)作为负对照。
预处理原始表后,我们过滤掉超过80%的时间点为零值的序列。最终,表S1和表S3包含4770对序列,其中10对时间序列具有真正的相关性。表S2共包含4366对序列,其中包含6对真相关时间序列,表S4共包含4285对序列,其中包含5对真相关时间序列。然后,我们将这些序列数据输入到每个LSA分析软件工具中,以计算协变量之间的成对LS分数。

Note: Tables S1-S10 can be obtained at http://github.com/labxscut/lsareview.
我们使用eLSA、fastLSA、MBBLSA和DDLSA工具对两物种Lotka-Volterra (TLV)数据集进行了基准测试。每种工具的性能在图2中由受试者工作特征(ROC)曲线(pROC的R包)说明。曲线显示,eLSA理论(theo)、eLSA置换(perm)和DDLSA方法在测试中普遍优于其他方法。具体而言,与所有其他方法相比,eLSA (theo)方法显示出最高的总体准确性。此外,我们用模拟多物种Lotka-Volterra关系的数据评估了这些工具,并比较了所得的ROC曲线,如补充表S1和图S1所示。我们观察到DDLSA方法在这些测试中产生了最好的性能。
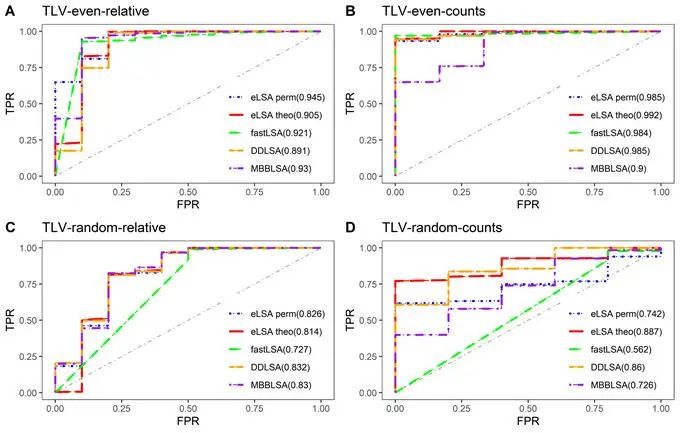
图2.四种LSA软件工具及其实现的p值近似方法的受试者工作特征(ROC)曲线。
图A-D显示了不同数据模拟的模型下的eLSA、fastLSA、DDLSA和MBBLSA的曲线下面积(AUC)得分。蓝色曲线表示置换的eLSA(eLSA-perm),红色曲线表示理论近似的eLSA(eLSA-theo)。同时,fastLSA(绿色)和DDLSA(橙色)是理论的,MBBLSA(紫色)是基于置换的近似。关于这些方法的进一步细节可以在正文中找到。
此外,表4显示了所有被测试软件工具正确识别的序列对的排序。表中n为工具计算的OTU对总数,准确列为工具正确识别的OTU对数。在这些工具中,我们观察到eLSA (theo)方法表现出最好的性能,在整体精度上排名第一。
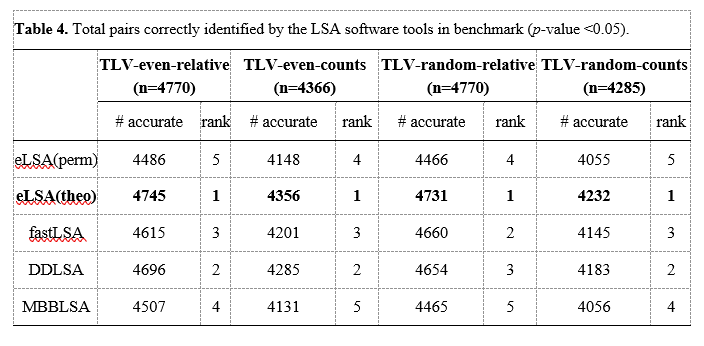
我们还从基准测试的总运行时间(以分钟为单位)方面比较了四种LSA软件工具的计算效率(见表5)。结果表明,所有理论方法eLSA (theo)、fastLSA和DDLSA在计算效率方面都表现出良好的性能,比置换方法的速度加快了数百倍以上。当处理数百个协变量(OTU)时,他们在几秒钟内完成了分析。我们的经验表明,使用当前的单处理器个人计算机,基于理论的方法可以扩展到数千个协变量,而排列方法则变得太慢而无法完成。总体而言,基准测试表明,考虑到总体准确性和效率,eLSA (theo)是最推荐的LSA分析选择。

局部趋势分析及统计学意义
本地趋势序列和本地趋势分析
一种相关的LSA分析技术-局部趋势分析(LTA)也在eLSA中实现,用于依赖时间的趋势关联挖掘。最近的研究表明,沿时间轴增加、稳定或减少趋势的相似性可以作为关联的有力指标。为了解决这个问题,趋势序列分析因此被发展用来分析转换后的序列。Ji等[15]探索了这一方法,将基因表达谱连续n个时间点的趋势编码为代表基因表达水平局部上升、不变、下降趋势的n−1个时间点序列。他们使用一种穷举搜索算法来分析转换后的序列,以确定基因之间特定长度或时间跨度可能存在的局部关联。后来,He等人[14]更新了分析,采用与LSA相同的动态规划算法和置换p值方法,计算相似度得分并评估统计显著性。因此,我们将LSA技术应用于变换后的趋势序列时称为局部趋势分析(local trend analysis, LTA),将其对应的相似性度量称为局部趋势(local trend, LT)得分。
在LTA中,第一步是使用变化的趋势字母表𝛴离散序列,它反映了一组感兴趣的符号,代表了状态的不同方向变化。通常,使用两个字母的字母表,如Σ={-1,1}表示趋势下降和趋势上升的状态,或者使用三个字母的字母表,如Σ={-1,0,1}表示趋势下降、不变和趋势上升的状态。离散成更多的字母也是可以实现的。

其中t≥0为表示变化趋势的阈值。
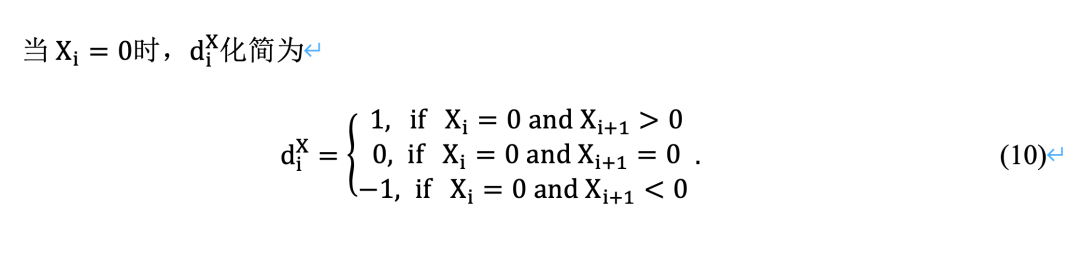
局部趋势评分的统计学意义
与LSA类似,LTA中LT得分的统计显著性理论是必要的。变换后得到的趋势序列不再是独立同分布的。如图3所示,它具有一个依赖结构,其中依赖关系随着时间的推移而减少。Xia等人[16]证明了一阶马尔可夫链在实践中可以很好地近似所得到的相关趋势序列:
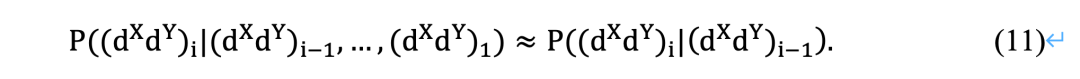
因此,LTA决定的LT分数S与LS分数相同,除了它代表了类马尔可夫随机变量之和的随机游走偏移的最大范围。

Xia等人关于LS分数的理论可以推广到Markov随机变量来近似LT分数的p值。Daudin等人[87]探讨了一阶马尔可夫链的最大累积和的分布,其值取自R的有限子集。设𝜑为这种马尔可夫随机变量Zi,i = 1,2,· · ·的平稳分布,其中Eφ (Z1 )=0且
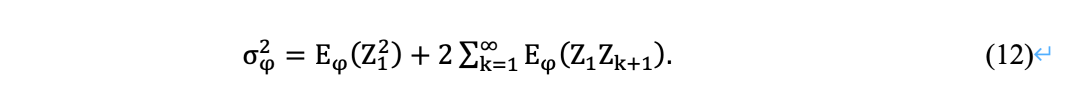
因此,遵循等式(6),LT评分的p值近似公式为:
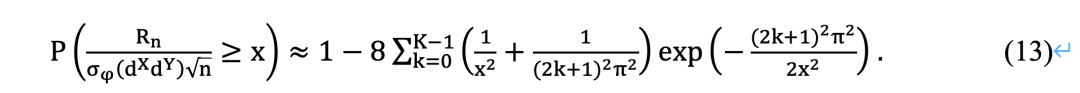


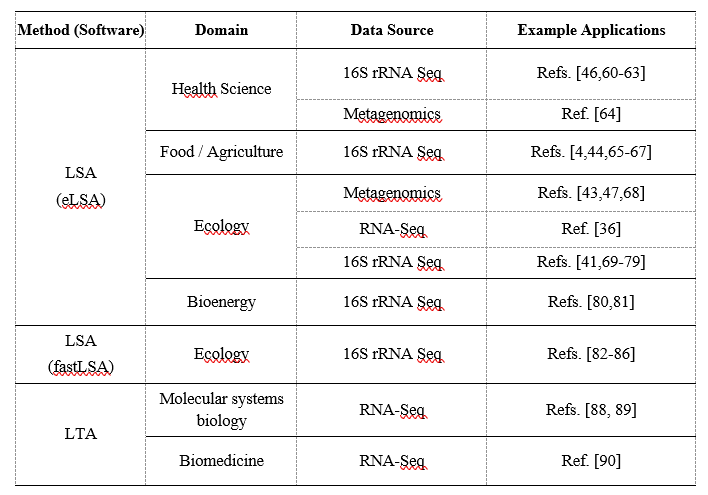
最后,表6总结了LSA和LTA软件工具及其在各种数据领域和专业中的实际应用,旨在为从业者提供这些工具广泛采用的方便概述。
- 讨论与结论 -
局部关联分析方法,如局部相似性和趋势分析(LSA和LTA),是识别生物时间序列数据中时间相关关联的有力工具。这些方法被广泛应用于假设生成,以确定生物系统中最相关的相互作用。通常采用标准的动态规划算法来寻找最优的时间序列对齐并计算局部相似度和趋势分数。理论统计显著性近似的进步解决了研究人员面临的常见计算瓶颈,特别是在分析大规模NGS数据集时,将LSA和LTA方法置于随机漫步的共同概念框架下进行理论研究。然而,在局部关联存在差距,全面比较数万个或甚至更多的变量,或分析局部关联涉及三个或更多的辅助因素的情况下,计算和理论上的挑战仍然存在。进一步发展新的算法和统计理论将使这些分析在未来成为可能。
目前可以使用多种软件工具来执行LSA。表2对eLSA、fastLSA、LocSim、MBBLSA和DDLSA这五种工具的特性进行了清晰的比较。虽然旧的R工具LocSim仅限于p值的排列过程,但最近的Python和c++实现,即eLSA,支持局部相似性和趋势分析,并提供p值的置换和理论近似。eLSA特别适用于基于NGS的大型数据集。eLSA还支持具有多个汇总函数和bootstrap置信区间的复制序列。最近发展的MBBLSA和DDLSA方法进一步改进了p值的置换和理论近似。此外,另一个c++实现fastLSA用p值的准确性来换取更快的计算速度。
本基准研究表明,无论在稀疏数据场景还是密集数据场景下,eLSA都是时间序列数据分析的首选软件工具。Weiss等人[28]也推荐eLSA和MIC[25]作为延迟D为零时密集截面数据的最佳分析方法。他们强调了LSA的鲁棒性(正如在eLSA中实现的那样),而不考虑数据集的分布和稀疏性。我们建议读者参考文献[28]对其中的综合评价有更深入的了解。此外,文献[29]对研究微生物群落网络的各种分析方法进行了总结。
要点
本文对局部相似性分析(LSA)进行了全面的回顾和分析,包括算法、统计理论和软件工具。
比较了各种软件工具的准确性和效率,以帮助读者选择最合适的工具。
References
1.Caporaso JG, Lauber CL, Costello EK et al. Moving pictures of the human microbiome. Genome Biol 2011;12:R50.
2.Cram JA, Xia LC, Needham DM et al. Cross-depth analysis of marine bacterial networks suggests downward propagation of temporal changes. ISME J 2015;9:2573-86.
3.Steele JA, Countway PD, Xia L et al. Marine bacterial, archaeal and protistan association networks reveal ecological linkages. ISME J 2011;5:1414-25.
4.Shade A, McManus PS, Handelsman J. Unexpected diversity during community succession in the apple flower microbiome. mBio 2013;4:e00602-12.
5.Cho RJ, Campbell MJ, Winzeler EA et al. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell 1998;2:65-73.
6.Spellman PT, Sherlock G, Zhang MQ et al. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell 1998;9:3273-97.
7.Amar D, Yekutieli D, Maron-Katz A et al. A hierarchical Bayesian model for flexible module discovery in three-way time-series data. Bioinformatics 2015;31:i17-26.
8.Vaisvaser S, Lin T, Admon R et al. Neural traces of stress: cortisol related sustained enhancement of amygdala-hippocampal functional connectivity. Front Hum Neurosci 2013;7:313.
9.Li KC. Genome-wide coexpression dynamics: theory and application. Proc Natl Acad Sci U S A 2002;99:16875-80.
10.Qian J, Dolled-Filhart M, Lin J et al. Beyond synexpression relationships: local clustering of time-shifted and inverted gene expression profiles identifies new. biologically relevant interactions, J Mol Biol 2001;314:1053-66.
11.Xia LC, Ai D, Cram J et al. Efficient statistical significance approximation for local similarity analysis of high-throughput time series data. Bioinformatics 2013;29:230-7.
12.Xia LC, Steele JA, Cram JA et al. Extended local similarity analysis (eLSA) of microbial community and other time series data with replicates. BMC Syst Biol 2011;5:S15.
13.Ruan Q, Dutta D, Schwalbach MS et al. Local similarity analysis reveals unique associations among marine bacterioplankton species and environmental factors. Bioinformatics 2006;22:2532-8.
14.He F, Zeng AP. In search of functional association from time-series microarray data based on the change trend and level of gene expression. BMC Bioinformatics 2006;7:69.
15.Ji L, Tan KL. Identifying time-lagged gene clusters using gene expression data. Bioinformatics 2005;21:509-16.
16.Xia LC, Ai D, Cram JA et al. Statistical significance approximation in local trend analysis of high-throughput time-series data using the theory of Markov chains. BMC Bioinformatics 2015;16:301.
17.Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol 1981;147:195-7.
18.Vasily R, Alexandr S, Natalia Z et al. A novel approach to local similarity of protein binding sites substantially improves computational drug design results. Proteins 2007;69:349-57.
19.Tachibana H, Inoue Y, Kanehisa T et al. Local Similarity in the Amino Acid Sequence between the Non-Catalytic Region of Rous Sarcoma Virus Oncogene Product p60v-src and Intermediate Filament Proteins. J Biochem 2008;104:869-72.
20.Raptis A. Local similarity transformations for the boundary layer flow through a homogeneous porous medium by the presence of heat transfer. Int Commun Heat Mass 2000;27:739-43.
21.Lai Y, Wu B, Chen L et al. A statistical method for identifying differential gene-gene co-expression patterns. Bioinformatics 2004;20:3146-55.
22.Li KC, Liu CT, Sun W et al. A system for enhancing genome-wide coexpression dynamics study. Proc Natl Acad Sci U S A 2004;101:15561-6.
23.Li X, Rao S, Jiang W et al. Discovery of Time-Delayed Gene Regulatory Networks based on temporal gene expression profiling. BMC Bioinformatics 2006;7:26.
24.Wang L, Liu S, Ding Y et al. Meta-analytic framework for liquid association. Bioinformatics 2017;33:2140-7.
25.Reshef DN, Reshef YA, Finucane HK et al. Detecting novel associations in large data sets. Science 2011;334:1518-24.
26.Ai D, Li X, Liu G et al. Constructing the microbial association network from large-scale time series data using granger causality. Genes 2019;10:216.
27.Ai D, Li X, Pan H et al. Explore mediated co-varying dynamics in microbial community using integrated local similarity and liquid association analysis. BMC Genomics 2019;20:185.
28.Weiss S, Van Treuren W, Lozupone C et al. Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. ISME J 2016;10:1669-81.
29.Matchado MS, Lauber M, Reitmeier S et al. Network analysis methods for studying microbial communities: A mini review. Comput Struct Biotechnol J 2021;19:2687-2698.
30.Durno WE, Hanson NW, Konwar KM et al. Expanding the boundaries of local similarity analysis. BMC Genomics 2013;14 Suppl 1:S3.
31.Zhang F, Shan A and Luan Y. A novel method to accurately calculate statistical significance of local similarity analysis for high-throughput time series. Stat Appl Genet Mol Biol 2018;17:20180019.
32.Zhang F, Sun F, Luan Y. Statistical significance approximation for local similarity analysis of dependent time series data. BMC Bioinformatics 2019;20:53.
33.Wang YXR, Liu K, Theusch E et al. Generalized correlation measure using count statistics for gene expression data with ordered samples. Bioinformatics 2018;34:617-624.
34.Tackmann J, Matias Rodrigues JF, von Mering C. Rapid inference of direct interactions in large-Scale Ecological Networks from Heterogeneous Microbial Sequencing Data. Cell Syst 2019;9:286-96.
35.Liu L, Yang J, Lv H et al. Synchronous dynamics and correlations between bacteria and phytoplankton in a subtropical drinking water reservoir. FEMS Microbiol Ecol 2014;90:126-38.
36.Thiriet-Rupert S, Carrier G, Trottier C et al. Identification of transcription factors involved in the phenotype of a domesticated oleaginous microalgae strain of Tisochrysis lutea. Algal Res 2018;30:59-72.
37.Lee YY, Lee SY, Lee SD et al. Seasonal dynamics of bacterial community structure in diesel oil-contaminated soil cultivated with tall fescue (Festuca arundinacea). Int J Environ Res Public Health 2022;19:4629.
38.Parada AE, Fuhrman JA. Marine archaeal dynamics and interactions with the microbial community over 5 years from surface to seafloor. ISME J 2017;11:2510-25.
39.Jones AC, Hambright KD, Caron DA. Ecological patterns among bacteria and microbial eukaryotes derived from network analyses in a Low-Salinity Lake. Microb Ecol 2018;75:917-29.
40.Liang Z, Xu G, Shi J et al. Sludge digestibility and functionally active microorganisms in methanogenic sludge digesters revealed by E. coli-fed digestion and microbial source tracking. Environ Res 2021, 193:110539.
41.Needham DM, Sachdeva R, Fuhrman JA. Ecological dynamics and co-occurrence among marine phytoplankton, bacteria and myoviruses shows microdiversity matters. ISME J 2017;11:1614-29.
42.Needham DM, Fuhrman JA. Pronounced daily succession of phytoplankton, archaea and bacteria following a spring bloom. Nat Microbiol 2016;1:16005.
43.Roux S, Chan LK, Egan R et al. Ecogenomics of virophages and their giant virus hosts assessed through time series metagenomics. Nat Commun 2017;8:858.
44.Wang H, Sangwan N, Li HY et al. The antibiotic resistome of swine manure is significantly altered by association with the Musca domestica larvae gut microbiome. ISME J 2017;11:100-11.
45.Posch T, Eugster B, Pomati F et al. Network of interactions between ciliates and phytoplankton during spring. Front Microbiol 2015;6:1289.
46.Džunková M, Martinez-Martinez D, Gardlík R et al. Oxidative stress in the oral cavity is driven by individual-specific bacterial communities. NPJ Biofilms and Microbiomes 2018;4:29.
47.Wang Y, Ye J, Ju F et al. Successional dynamics and alternative stable states in a saline activated sludge microbial community over 9 years. Microbiome 2021;9:199.
48.Shan A, Zhang F, Luan Y. Efficient approximation of statistical significance in local trend analysis of dependent time series. Front Genet 2022;13:729011.
49.Lee ML, Kuo FC, Whitmore GA et al. Importance of replication in microarray gene expression studies: statistical methods and evidence from repetitive cDNA hybridizations. Proc Natl Acad Sci U S A 2000;97:9834-9.
50.Nguyen TT, Almon RR, DuBois DC et al. Importance of replication in analyzing time-series gene expression data: corticosteroid dynamics and circadian patterns in rat liver. BMC Bioinformatics 2010;11:279.
51.Zhu D, Li Y, Li H. Multivariate correlation estimator for inferring functional relationships from replicated genome-wide data. Bioinformatics 2007;23:2298-305.
52.Yao J, Chang C, Salmi ML et al. Genome-scale cluster analysis of replicated microarrays using shrinkage correlation coefficient. BMC Bioinformatics 2008;9:288.
53.Littell RC, Pendergast J, Natarajan R. Modelling covariance structure in the analysis of repeated measures data. Stat Med 2000;19:1793-819.
54.Leroy AM, Rousseeuw PJ. Robust regression and outlier detection. John Wiley & Sons,2005.
55.Hoaglin DC, Mosteller F, Tukey JW. Understanding robust and exploratory data analysis. Wiley series in probability and mathematical statistics,1983.
56.Venables WN, Ripley BD. Modern applied statistics with S. Fourth edition. New York, NY: Springer-Verlag; 2002.
57.Sherman M, Speed Jr FM, Speed, FM. Analysis of tidal data via the blockwise bootstrap. J Appl Stat 1998; 25:333-340.
58.Feller W. The Asymptotic distribution of the range of sums of independent random variables. Ann Math Statist 1951;22:427-32.
59.Andrews D. Heteroskedasticity and autocorrelation consistent covariance matrix estimation. Econometrica 1991;59:817-58.
60.Seekatz AM, Panda A, Rasko DA et al. Differential Response of the Cynomolgus Macaque Gut Microbiota to Shigella Infection. PLoS ONE 2013;8:e64212.
61.Sun J, Liao XP, D'Souza AW et al. Environmental remodeling of human gut microbiota and antibiotic resistome in livestock farms. Nat Commun 2020;11:1427.
62.Zheng W, Huyan J, Tian Z et al. Clinical class 1 integron-integrase gene - A promising indicator to monitor the abundance and elimination of antibiotic resistance genes in an urban wastewater treatment plant. Environ Int 2020;135:105372.
63.Copeland E, Leonard K, Carney R et al. Chronic Rhinosinusitis: Potential Role of Microbial Dysbiosis and Recommendations for Sampling Sites. Front Cell Infect Microbio 2018;8:57.
64.Xu J, Wang H, Xu R et al. The diurnal fluctuation of colonic antibiotic resistome is correlated with nutrient substrates in a pig model. Sci Total Environ 2023;891:164692.
65.Jiang CL, Jin WZ, Tao XH et al. Black soldier fly larvae (Hermetia illucens) strengthen the metabolic function of food waste biodegradation by gut microbiome. Microb Biotechnol 2019;12:528-543.
66.Simons AL, Churches N, Nuzhdin S. High turnover of faecal microbiome from algal feedstock experimental manipulations in the Pacific oyster (Crassostrea gigas). Microb Biotechnol 2018;11:848-858.
67.Garcia J, Gannett M, Wei L et al. Selection pressure on the rhizosphere microbiome can alter nitrogen use efficiency and seed yield in Brassica rapa. Commun Biol 2022;5:959.
68.Ki BM, Ryu HW, Cho KS. Extended local similarity analysis (eLSA) reveals unique associations between bacterial community structure and odor emission during pig carcasses decomposition. J Environ Sci Health A Tox Hazard Subst Environ Eng 2018;53:718-727.
69.Pollet T, Berdjeb L, Garnier C et al. Prokaryotic community successions and interactions in marine biofilms: the key role of Flavobacteriia. FEMS Microbiol Ecol 2018;94.
70.Chow CE, Sachdeva R, Cram JA et al. Temporal variability and coherence of euphotic zone bacterial communities over a decade in the Southern California Bight. ISME J 2013;7:2259-73.
71.Ju F, Zhang T. Bacterial assembly and temporal dynamics in activated sludge of a full-scale municipal wastewater treatment plant. ISME J 2015;9:683-95.
72.Kankan Z,Haodan Y,Ran X et al. The only constant is change: Endogenous circadian rhythms of soil microbial activities. Soil Biol Biochem. 2022;173:108805.
73.Lee YY, Seo Y, Ha M et al. Evaluation of rhizoremediation and methane emission in diesel-contaminated soil cultivated with tall fescue (Festuca arundinacea). Environmental Research 2021;194:110606.
74.Thomas F, Cébron A. Short-Term Rhizosphere Effect on Available Carbon Sources, Phenanthrene Degradation, and Active Microbiome in an Aged-Contaminated Industrial Soil. Front Microbiol 2016;7:92.
75.Lee YY, Lee SY, Cho KS. Phytoremediation and bacterial community dynamics of diesel-and heavy metal-contaminated soil: Long-term monitoring on a pilot scale. International Biodeterioration & Biodegradation 2023;183:105642.
76.Lee YY, Choi H, Cho KS. Effects of carbon source, C/N ratio, nitrate, temperature, and pH on N2O emission and functional denitrifying genes during heterotrophic denitrification. J Environ Sci Health A Tox Hazard Subst Environ Eng 2019;54:16-29.
77.Fletcher-Hoppe C, Yeh YC, Raut, Y et al. Symbiotic UCYN-A strains co-occurred with El Niño, relaxed upwelling, and varied eukaryotes over 10 years off Southern California. ISME COMMUN 2023;3:63.
78.Kwon JH, Park HJ, Lee YY et al. Evaluation of denitrification performance and bacterial community of a sequencing batch reactor under intermittent aeration. J Environ Sci Health A Tox Hazard Subst Environ Eng 2020;55:179-192.
79.Carini P, Delgado-Baquerizo M, Hinckley ES et al. Effects of Spatial Variability and Relic DNA Removal on the Detection of Temporal Dynamics in Soil Microbial Communities. mBio 2020;11:e02776-19.
80.Kim TG, Yun J, Cho KS. The close relation between Lactococcus and Methanosaeta is a keystone for stable methane production from molasses wastewater in a UASB reactor. Appl Microbiol Biotechnol 2015;99:8271-83.
81.Lee YY, Kim TG, Cho KS. Effects of proton exchange membrane on the performance and microbial community composition of air-cathode microbial fuel cells. J Biotechnol 2015;211:130-7.
82.Steffen K, Indraningra, AAG, Erngren I et al. Oceanographic setting influences the prokaryotic community and metabolome in deep-sea sponges. Sci Rep 2022;12:3356.
83.Jang J, Park J, Hwang CY et al. Abundance and diversity of antibiotic resistance genes and bacterial communities in the western Pacific and Southern Oceans. Sci Total Environ 2022;822:153360.
84.Zhuang Y, Chai J, Cui K et al. Longitudinal Investigation of the Gut Microbiota in Goat Kids from Birth to Postweaning. Microorganisms 2020;8:1111.
85.Bergk Pinto B, Maccario L, Dommergue A et al. Do Organic Substrates Drive Microbial Community Interactions in Arctic Snow? Front Microbiol 2019;10:2492.
86.Auladell A, Sánchez P, Sánchez O et al. Long-term seasonal and interannual variability of marine aerobic anoxygenic photoheterotrophic bacteria. ISME J 2019;13:1975-87.
87.Daudin JJ, Etienne MP, Vallois P. Asymptotic behavior of the local score of independent and identically distributed random sequences. Stoch Proc Appl 2003;107:1-28.
88.He F, Chen H, Probst-Kepper M et al. PLAU inferred from a correlation network is critical for suppressor function of regulatory T cells. Mol Syst Biol 2012;8:624.
89.Gonçalves PJ, Aires SR, Francisco PA et al. Regulatory Snapshots: integrative mining of regulatory modules from expression time series and regulatory networks. PLoS ONE 2017;7:e35977.
90.Sudhakar P, Reck M, Wang W et al. Construction and verification of the transcriptional regulatory response network of Streptococcus mutans upon treatment with the biofilm inhibitor carolacton. BMC Genomics 2014;15:362.
参考文献
Ai, D., Chen, L., Xie, J., Cheng, L., Zhang, F., Luan, Y., Li, Y., Hou, S., Sun, F., & Xia, L. C. (2023). Identifying local associations in biological time series: algorithms, statistical significance, and applications. Briefings in Bioinformatics, 24(6). https://doi.org/10.1093/bib/bbad390
- 作者简介 -
第一作者

北京科技大学
艾冬梅
教授
艾冬梅,北京科技大学统计系教授,博士生导师。1998.02-至今,在北京科技大学数理学院工作。2011.10-2012.10在美国南加州大学生物科学系,做访问学者。主要研究方向:生物与医学大数据智能计算,围绕微生物宏基因组,基因组学,免疫组学等组学,运用机器学习和统计算法模型,探究生物标记物致病机理。主持或参与国家自然科学基金面上项目、国家“863”计划等多项,在Bioinformatics、BIB等杂志发表论文60余篇。担任北京人工智能学会常务理事,中国人工智能学会、自动化学会、计算机学会生物信息相关专业委员会委员。
合作作者

南加州大学
孙丰珠
教授
孙丰珠博士是南加州大学(USC)的定量和计算生物学教授,博导。他在山东大学获得数学专业学士学位,在北京大学获得概率与统计学专业硕士,在南加州大学获得应用数学博士。他是美国科学促进会(AAAS)、美国统计协会(ASA)和数学统计研究所(IMS)的Fellow。2012年,他获得了南加州大学教务长梅隆导师奖,2017年,他因在研究、教学和服务方面的卓越表现,获得南加州大学Dornsife学院高级劳本海默奖。根据谷歌学者的数据,他发表了200多篇论文,引用次数超过13000次。
通讯作者

华南理工大学
夏立
教授
夏立,国家级青年人才 (2020)、美国癌症协会博士后会士(2019)、美国癌症研究会培训学者奖(2018)。一名硅谷海归的网球、跑步和科技爱好者。研究方向:统计学、计算机科学、机器学习、人工智能与生物医学大数据和精准医疗相交叉方向。在《自然-医学》、《核酸研究》、《生物信息学》等国际顶级期刊上发表论文37篇,包括3篇ESI高引论文,11次应邀在美国遗传学年会,美国癌症研究会年会,国际阿氏症协会年会等顶级会议作报告。主持国家海外高层次人才基金,美国癌症信息学创新基金重大项目,美国癌症协会博士后基金等3项。
宏基因组推荐
猜你喜欢
iMeta高引文章 fastp 复杂热图 ggtree 绘图imageGP 网络iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文,跳转最新文章目录阅读

















 3547
3547

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









