







数据整理是数据分析的前提,而数据的正确提取则是数据整理的前提。
这次曾老师交代了一个作业,希望对两个GEO转录组的数据(GSE216943, GSE236049)进行差异分析。
从GEO数据库中可以发现,这两个数据集都需要下载研究者整理好的原始数据进行整合。
GSE216943数据集:

GSE236049数据集:

针对GSE216943数据集,我仅下载了count数据,然后循环读取(如下是其中一个count文件的样式,里边的列名是id和样本名称)

然而针对GSE236049数据集,我发现研究者把文件整理的很棒,但是对于生信菜鸟的我有点不知道怎么合理的读取并处理信息(如下是其中一个文件,里边有基因名称,FPKM,TPM等值)。

没办法,只好去向曾老师求助了hhhh 。曾老师立刻发给我了一份其他高手处理的流程6666666。最后这位高手的代码也解决了困扰我的问题。
为此,我也顺便把多种提取转录组数据的方法整理了一下,以供大家参考。
转录组数据提取技巧汇总
提取数据前,我们都需要注意获得的数据是count还是tpm数据(首选count数据),只有这两种格式的可以进行差异分析。其他类型的数据如log2cpm,fpkm,rpkm在进行差异表达分析时,需要重新修正为count或者tpm数据。
情况1.可以通过get_count_txt函数获得count数据
library(tinyarray)
get_count_txt("GSE242333")
exp = read.table("GSE242333_raw_counts_GRCh38.p13_NCBI.tsv.gz",check.names = F,row.names = 1,header = T)
复制网址就可以自动下载数据,之后读取即可。


情况2. 需自行下载数据,数据已经被研究者整合好了
#下载后直接读取
#GSE121949
a = read.delim("GSE121949_RNAseq_Raw_and_RPKM.txt")
#GSE235017
dat = data.table::fread("GSE235017_ref_trans_full_table.txt",check.names = F, header = T)


情况3. 原始数据可直接循环读取合并
file_directory = "~/desktop/training/GSE216943_RAW"
fs=list.files(file_directory)
dat = do.call(cbind,lapply(fs, function(x){
read.table(file.path(file_directory,x),
header = T,sep = '\t',row.names = 1,quote = "")
}))
dat[1:4,1:4]
GSE216943数据集中的其中一个样本文件

情况4. Affymetrix的CEL文件读取合并及处理
#BiocManager::install("affy")
library(affy)
dir_cels='~/Desktop/GSE29330/GSE29330_RAW'
affy_data = ReadAffy(celfile.path=dir_cels)
eSet = rma(affy_data) #rma函数去normalization
##要进行过滤
calls = mas5calls(affy_data) # get PMA calls
calls = exprs(calls)
absent = rowSums(calls == 'A') # how may samples are each gene 'absent' in all samples
absent = which(absent == ncol(calls)) # which genes are 'absent' in all samples
eSet = eSet[-absent,] # filters out the genes 'absent' in all samples
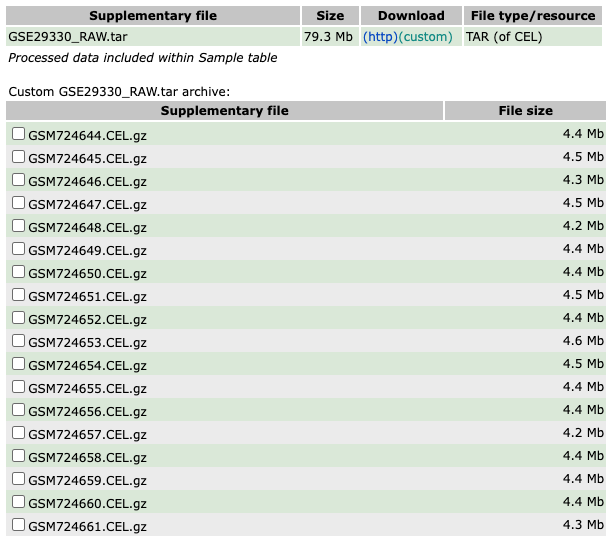
情况5. 数据需要自行下载,并且文件内容中没有样本信息
#代码来自 艾迪
# 读取文件信息
exp.dirs<-list.files("data/GSE236049_RAW/",full.names = T) #读取完整文件及子文件路径信息
exp.names<-list.files("data/GSE236049_RAW/") #读取子文件的名称信息(区别在于full.name是否为T)
#提取样本名称信息
#需要提取的名称信息应根据实际情况而定
DirInfor <- stringr::str_split(exp.names,"_",n = Inf, simplify = T)%>%
as.data.frame()
#清洗raw文件并合并
expList <- mapply(function(sampleDir, sampleName) {
df1 <- data.table::fread(sampleDir, data.table = F) %>%
dplyr::select("Gene Name", "FPKM", "TPM") %>% #选择需要的列
dplyr::rename(gene = "Gene Name")%>% #重命名列
dplyr::filter(!is.na(gene))%>% #过滤掉基因名为空的行
dplyr::filter(!gene == "-")%>% #过滤掉基因名为 "-" 的行
dplyr::filter(!duplicated(gene)) #移除基因名重复的行
names(df1)[2:3] <- paste0(sampleName, "_", names(df1)[2:3]) #修改列名
return(df1) }, #返回处理后的列表
sampleDir = exp.dirs, # 输入参数
sampleName = DirInfor$V1, # 输入参数
SIMPLIFY = F) #结果不简化为矩阵或数组,而是保持为列表
#获取FPKM/TPM数据
exps <- Reduce(function(x, y) merge(x, y, all = TRUE), expList)
#获取FPKM数据
fpkm <- exps%>%
tibble::as_tibble()%>%
tibble::column_to_rownames("gene")%>% #将tibble的gene列转换为行名,并从数据中移除这一列
dplyr::select(contains("FPKM"))%>% #选择包含FPKM的列
dplyr::rename_all(~ gsub("_FPKM", "", .))
#获取TPM数据
tpm <- exps%>%
tibble::as_tibble()%>%
tibble::column_to_rownames("gene")%>%
dplyr::select(contains("TPM"))%>%
dplyr::rename_all(~ gsub("_TPM", "", .))
#数据检查
tpm%>%colSums()%>%table()
类似于GSE236049数据集提供的原始数据样式

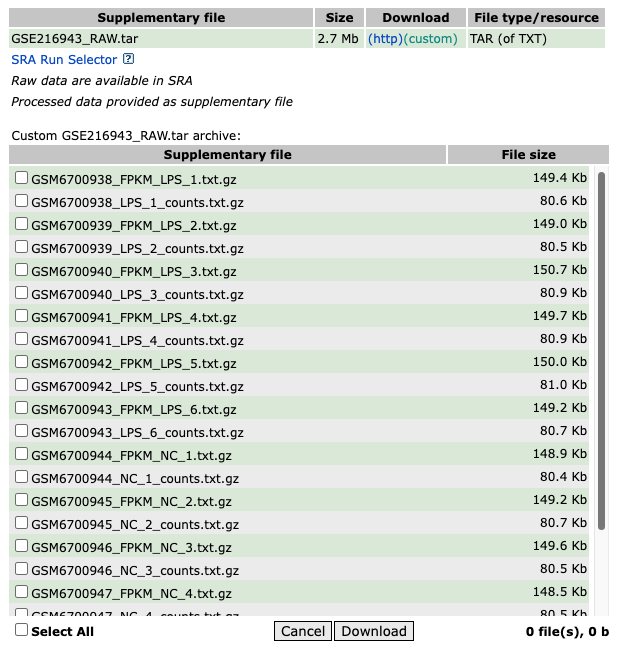
情况6. 如果样本量大,原始文件中存在不想提取的数据
#代码来自 艾迪
count.dirs<-list.files("data/GSE216943_RAW/","counts",full.names = T) #在目录下搜索包含count的文件
fpkm.dirs<-list.files("data/GSE216943_RAW/","FPKM",full.names = T)
#读取count数据
count <- lapply(count.dirs, function(x) df<-data.table::fread(x))%>%
as.data.frame()%>%
tibble::column_to_rownames("id")%>%
dplyr::select(contains("NC")|contains("LPS"))
#读取fpkm数据
fpkm<-lapply(fpkm.dirs, function(x) df<-data.table::fread(x))%>%
as.data.frame()%>%
tibble::column_to_rownames("id")%>%
dplyr::select(contains("NC")|contains("LPS"))
#fpkm转换成TPM
fpkmToTpm <- function(fpkm){(fpkm/sum(fpkm))*10^6} #定义转换函数
tpm <- apply(fpkm,2, fpkmToTpm) #2的含义是针对每个样本
#数据检查
tpm%>%colSums()%>%table()
类似于GSE216943数据集提供的原始数据样式,假设数据集中的样本量很大的时候,我们手动勾选下载会比较麻烦。
小结:数据提取也不是那么容易的哦~
致谢:感谢曾老师,艾迪,小洁老师以及生信技能树团队全体成员(部分代码来源:生信技能树马拉松和数据挖掘课程)。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(希望多多交流)。更多内容可关注公众号:生信方舟
- END -















 489
489











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









