







通常情况下,对两组或多组样本进行了不同处理/干预之后,研究者首先会进行同种细胞亚群处理前后的细胞数量的比较,但在单细胞分辨率时代之后,即使是同一个亚群中的不同细胞也应当看成不同的样本。
那么问题就来了,既然应当看做是不同的样本,那么干预之后假设同一种细胞亚群中的细胞数量产生了变化,这种变化是否还有借鉴和研究的价值(单从细胞数层面来看),笔者认为必然是有借鉴和研究的价值的。虽然确实是需要把不同细胞看做是不同类型的样本,但这种差异也还是不能掩盖它们在生物学上作为某一大类细胞的本身属性,而每一个细胞的异质性也应当认为是基于大类细胞的特征之下的子分类。
那么还有人可能会钻一下牛角尖,由于细胞亚群定义是基于先验知识,也就是人为的先根据已知的几个重要标记基因去定义细胞亚群,那么假设细胞亚群定义错了会不会导致最后的差异分析结果产生问题呢,这种情况也必然是存在的(主要存在于复杂的细胞亚群)。
因此有研究者就提出了miloR这种细胞亚群差异丰度的方法,这种方法有一个非常大的优势在于能够避免预先定义的聚类依赖性(预先定义的细胞亚群) 。
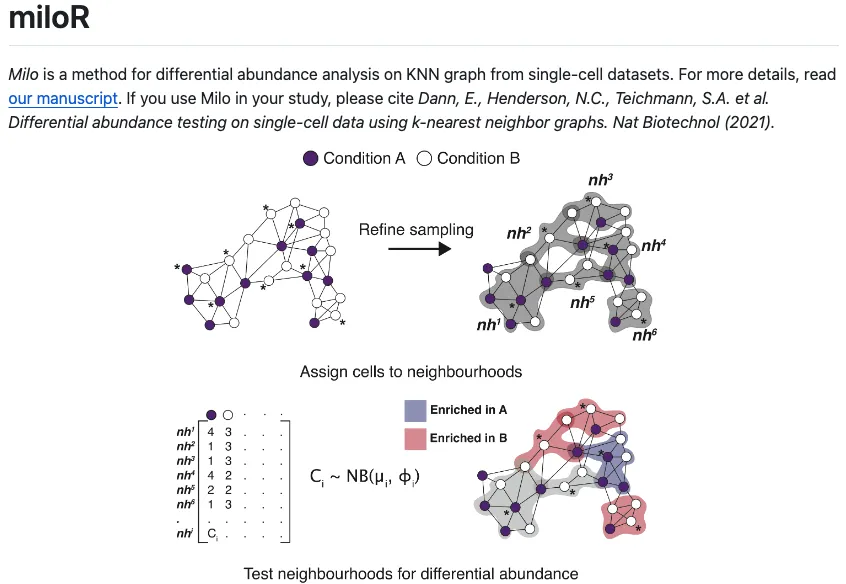
假设我们有一批包含了疾病和正常信息的单细胞测序数据,miloR先随机定义细胞中的细胞节点(数据点),然后通过K最近邻法(K-neareat neighbors)去识别所定义节点与周围的其他细胞数据点之间的邻近关系,找到跟这些节点最近的(比如欧几里得法)其他数据点(细胞) ,将这些具有邻近关系的细胞小群看做不同的群组,此时我们可以想象一下,每个群组中都会存在具有疾病和正常信息的许多细胞,接着再将这些群组中疾病与否的细胞数分别进行比较和汇总,最后可以得到基于某一大类细胞中内部细胞变化趋势的结果(比如是否倾向与疾病的方向)。
有了这个结果之后,无论是结合预先定义的细胞分群结果还是后续作为细胞分群结果的参考都有助于使用者更好的去了解疾病与否的不同细胞亚群的变化状态,因此使用miloR可以有效的作为单细胞水平上细胞差异分析的补充工具。
分析流程
1.导入
rm(list = ls())
V5_path = "/Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/seurat5/"
.libPaths(V5_path)
.libPaths()
library(stringr)
library(Seurat)
library(miloR)
library(scater)
library(scran)
library(dplyr)
library(patchwork)
library(qs)
library(BiocParallel)
register(MulticoreParam(workers = 4, progressbar = TRUE))
load("scRNA.Rdata")
2.数据预处理
# 提取两个样本
# check
table(scRNA$orig.ident)
# sample1 sample2 sample3 sample4 sample5 sample6
# 687 622 683 686 678 675
table(scRNA$celltype)
# CD4+ T-cells Fibroblasts B-cells CD8+ T-cells Neutrophils
# 1007 762 667 547 378
# Monocytes Adipocytes NK cells Endothelial cells
# 311 244 87 28
dat <- scRNA@meta.data
# 增加一下批次信息,转换成数字
dat$batch <- ifelse(grepl("sample2|sample4|sample6", dat$orig.ident), "1", "2")
# 增加一下分组信息,这里是随意编造的
dat$group <- ifelse(grepl("sample[1-3]", dat$orig.ident), "con", "treat")
scRNA@meta.data <- dat
3.构建miloR对象
# 由于需要输入SingleCellExperiment对象
# 因此先进行转化一下
scRNA_pre <- as.SingleCellExperiment(scRNA_pre)
# 构建miloR对象
scRNA_pre <- Milo(scRNA_pre)
scRNA_pre
# class: Milo
# dim: 24357 1309
# metadata(0):
# assays(3): counts logcounts scaledata
# rownames(24357): AL627309.1 AL627309.3 ... AF127936.2 AF127577.3
# rowData names(0):
# colnames(1309): AAAGAACAGTCTGTAC-1_1 AAAGGGCTCTCGCGTT-1_1 ...
# TTTGGAGAGTTCCGGC-1_2 TTTGTTGCAAGTTCGT-1_2
# colData names(10): orig.ident nCount_RNA ... celltype ident
# reducedDimNames(4): PCA HARMONY UMAP TSNE
# mainExpName: RNA
# altExpNames(0):
# nhoods dimensions(2): 1 1
# nhoodCounts dimensions(2): 1 1
# nhoodDistances dimension(1): 0
# graph names(0):
# nhoodIndex names(1): 0
# nhoodExpression dimension(2): 1 1
# nhoodReducedDim names(0):
# nhoodGraph names(0):
# nhoodAdjacency dimension(2): 1 1
4.构建K最邻图谱
# d值与降维时选定的pca值一致
# k值与FindNeighbors时选定的值一致
reducedDims(scRNA_pre)
# List of length 4
# names(4): PCA HARMONY UMAP TSNE
scRNA_pre <- buildGraph(scRNA_pre, k = 15, d = 15,reduced.dim = "PCA")
5.在 KNN 图上定义具有代表性的邻域
# 定义数据点的值(Prop)
# 对于小于3万细胞的数据集,Prop设为0.1
# 对于超过3万细胞的数据集,prop设为0.05。
scRNA_pre <- makeNhoods(scRNA_pre, prop = 0.1, k = 15, d = 15,
refined = TRUE, reduced_dims = "PCA")
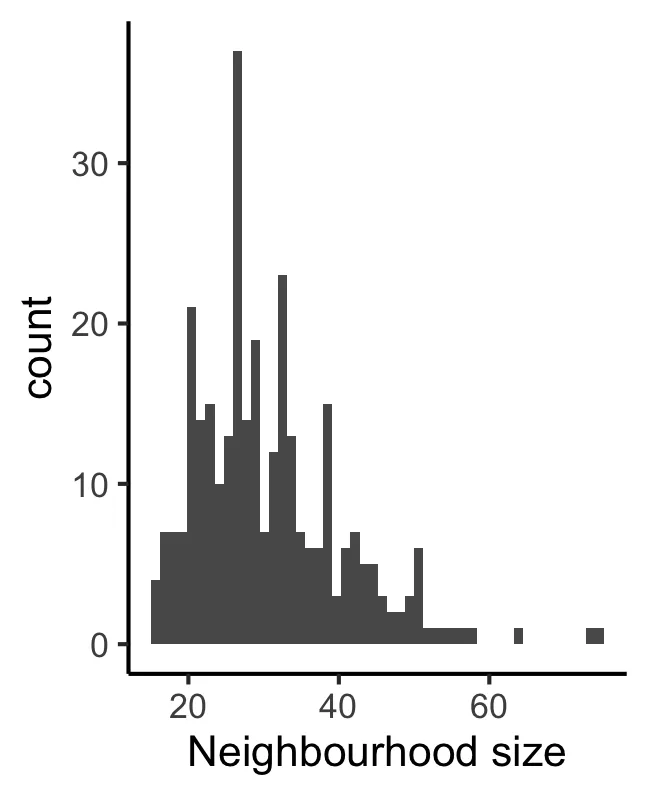
plotNhoodSizeHist(scRNA_pre)

这里定义的每个neighbourhood大小是不少于5个细胞/样本(笔者个人理解)
6.计算neighbourhoods中的细胞
scRNA <- countCells(scRNA,
meta.data = as.data.frame(colData(scRNA)),
sample="orig.ident")
# colData(scRNA_pre) 相当于代表了metadata的信息
# sample代表了样本数
head(nhoodCounts(scRNA))
# 这个步骤能够给scRNA_pre数据中增加一个n*m的matrix
# n代表不同的neighborhood数量,m代表实验样本
# 6 x 6 sparse Matrix of class "dgCMatrix"
# sample1 sample2 sample3 sample4 sample5 sample6
# 1 . 10 . 7 20 9
# 2 . 17 . 6 6 4
# 3 22 2 50 . . 1
# 4 20 2 18 1 1 7
# 5 2 6 6 7 1 5
# 6 5 9 3 . . 5
7.实验设计定义
# 笔者这里batch为批次信息
scRNA_design <- data.frame(colData(scRNA))[,c("orig.ident","group","batch")]
## 将批次信息转换为整数
scRNA_design$batch <- as.factor(scRNA_design$batch)
scRNA_design <- distinct(scRNA_design)
rownames(scRNA_design) <- scRNA_design$orig.ident
scRNA_design
# orig.ident group batch
# sample1 sample1 con 2
# sample2 sample2 con 1
# sample3 sample3 con 2
# sample4 sample4 treat 1
# sample5 sample5 treat 2
# sample6 sample6 treat 1
8.计算邻里连通性
# MiloR使用了由cydar引入的空间FDR校正,这一步可以校正邻里之间重叠的p值
scRNA <- calcNhoodDistance(scRNA, d=15, reduced.dim = "PCA")
9.检测结果
# 把批次和分组信息加到design中去
da_results <- testNhoods(scRNA,design = ~ batch + group, design.df = scRNA_design)
head(da_results)
# logFC logCPM F PValue FDR Nhood SpatialFDR
# 1 3.1406854 12.86784 1.053518712 0.3049667 0.7636565 1 0.9681699
# 2 1.5241056 12.40488 0.463991996 0.4959349 0.9371039 2 0.9681699
# 3 -3.9628982 12.75593 1.284826869 0.2572975 0.7258632 3 0.9681699
# 4 -1.0256918 12.35456 0.131264086 0.7172078 0.9382545 4 0.9681699
# 5 -0.1763181 12.06026 0.008314397 0.9273667 0.9649626 5 0.9681699
# 6 -2.5291223 11.67320 1.547069145 0.2138841 0.6816075 6 0.9681699
da_results %>%
arrange(SpatialFDR) %>%
head()
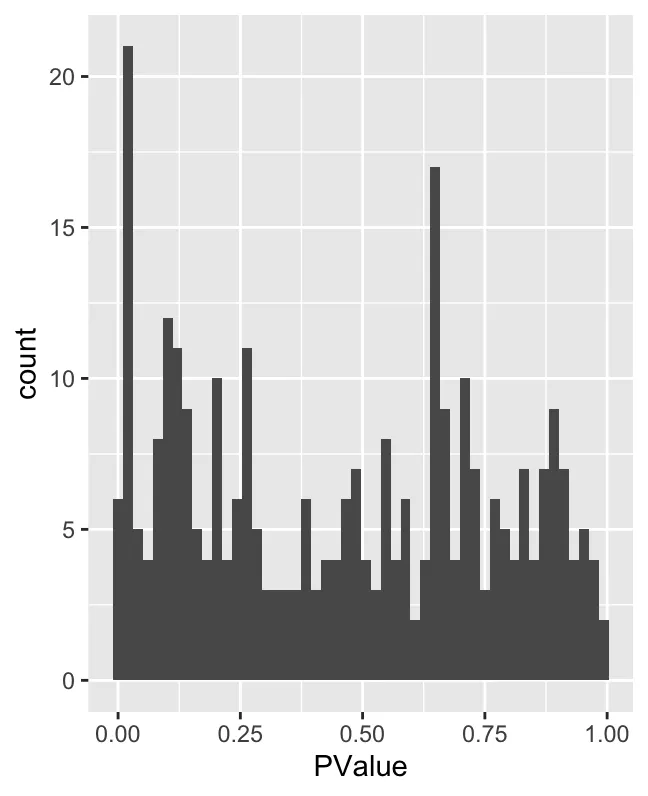
ggplot(da_results, aes(PValue)) + geom_histogram(bins=50)
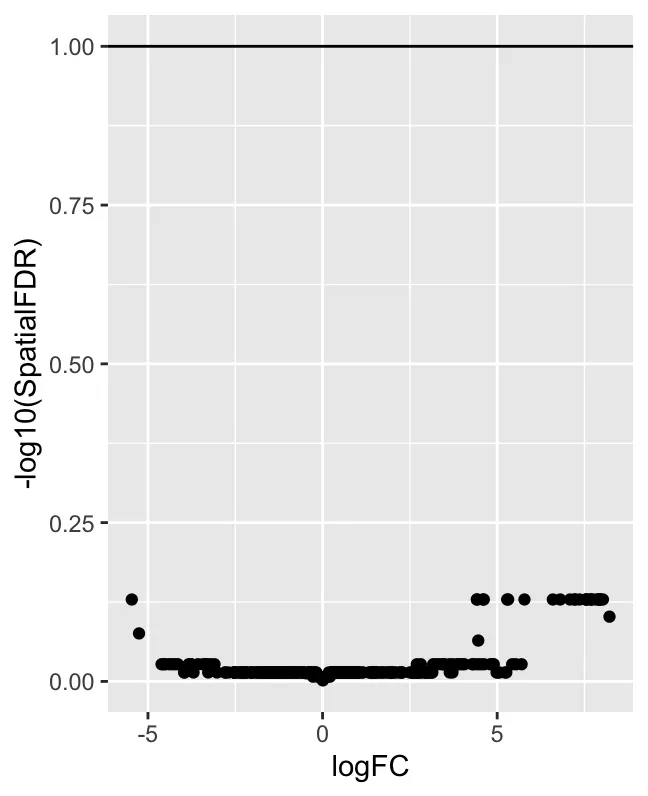
ggplot(da_results, aes(logFC, -log10(SpatialFDR))) +
geom_point() +
geom_hline(yintercept = 1) ## Mark significance threshold (10% FDR)
P-value 直方图:展示了 P 值的分布。通常,在差异丰度测试(例如 MiloR)中,P 值可以衡量每个邻域的统计显著性。如果一个 P 值接近 0,意味着该邻域在两个条件之间有显著差异。从图中可以看出,P 值接近 0 的邻域数量最多,但 P 值分布相对均匀。这表明大多数邻域没有很强的统计显著性,少数邻域具有较低的 P 值(即显著差异)
logFC vs -log10(SpatialFDR) 散点图:一个火山图样式的散点图,x 轴是 logFC,y 轴是 -log10(SpatialFDR)。 logFC 表示对数折叠变化,用来表示不同分组(如 treat 和 con)中某个邻域的相对丰度变化。正值表示该邻域在 treatment 组中富集,负值表示在 control 组中富集。SpatialFDR 是经过空间校正的FDR。通过绘制 -log10(SpatialFDR),该值越高,显著性越强(因为 FDR 越小)。绝大多数点都位于 y 轴较低的地方,表明这些邻域的 SpatialFDR 值较高(即不显著)。换句话说,测试发现的差异邻域很少,表明数据中没有强烈的差异丰度信号。
10.可视化
scRNA <- buildNhoodGraph(scRNA)
## Plot single-cell UMAP
umap_pl <- plotReducedDim(scRNA, dimred = "UMAP",
colour_by="group", text_by = "celltype",
text_size = 3, point_size=0.5) + guides(fill="none")
umap_pl
## Plot neighbourhood graph
nh_graph_pl <- plotNhoodGraphDA(scRNA, da_results,
layout="UMAP",alpha=0.9) #alpha默认0.1
umap_pl + nh_graph_pl +
plot_layout(guides="collect")
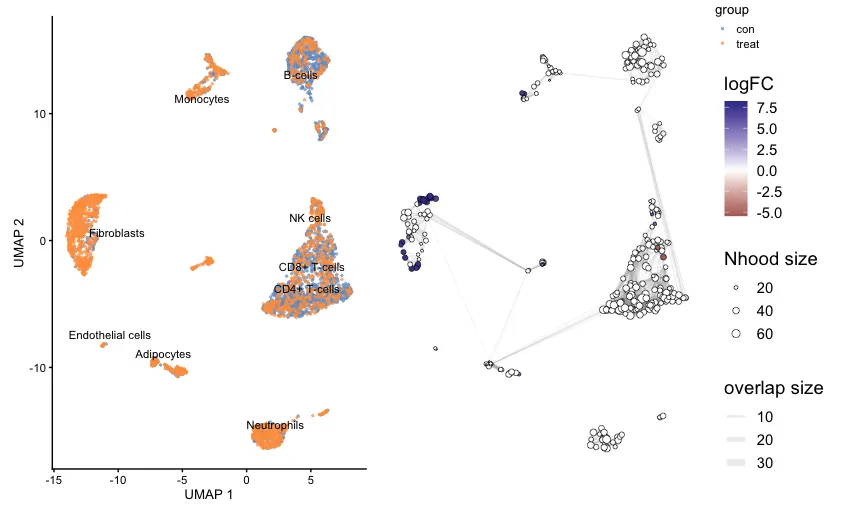
UMAP 图(左图):显示了不同细胞类型在两个实验组之间的分布。图中使用了两种颜色,蓝色(con) 和 橙色(treat),分别代表对照组和处理组。不同的细胞类型被明确标注(如 Fibroblasts、B-cells、Monocytes 等),并且可以看到处理组(橙色)和对照组(蓝色)之间的细胞分布。
邻域网络图(右图):右边的图是基于 MiloR 分析的邻域图。每个圈代表一个邻域(Nhood),邻域的大小与其中细胞的数量成比例。颜色梯度(从蓝色到红色)表示了 logFC(对数折叠变化):正的 logFC(蓝色)意味着该邻域中的细胞在对照组中富集,负的 logFC(红色)则表明该邻域中的细胞在处理组中富集。邻域之间的连线表示邻域之间的相似性或重叠,线条的厚度表示邻域之间的重叠度(overlap size),越厚的线表示两个邻域之间的细胞更为相似。
11.数据注释
da_results <- annotateNhoods(scRNA,
da_results,
coldata_col = "celltype")
head(da_results)
# logFC logCPM F PValue FDR Nhood SpatialFDR celltype celltype_fraction
# 1 3.1406854 12.86784 1.053518712 0.3049667 0.7636565 1 0.9681699 Neutrophils 1.0000000
# 2 1.5241056 12.40488 0.463991996 0.4959349 0.9371039 2 0.9681699 NK cells 1.0000000
# 3 -3.9628982 12.75593 1.284826869 0.2572975 0.7258632 3 0.9681699 CD4+ T-cells 1.0000000
# 4 -1.0256918 12.35456 0.131264086 0.7172078 0.9382545 4 0.9681699 CD4+ T-cells 1.0000000
# 5 -0.1763181 12.06026 0.008314397 0.9273667 0.9649626 5 0.9681699 Monocytes 1.0000000
# 6 -2.5291223 11.67320 1.547069145 0.2138841 0.6816075 6 0.9681699 CD4+ T-cells 0.9545455
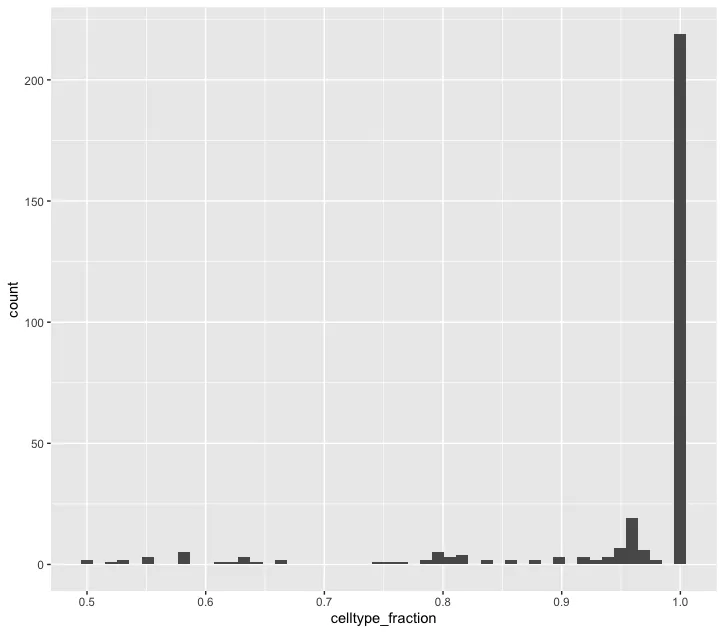
ggplot(da_results, aes(celltype_fraction)) + geom_histogram(bins=50)
str(da_results)
table(da_results$celltype)
range(da_results$SpatialFDR)
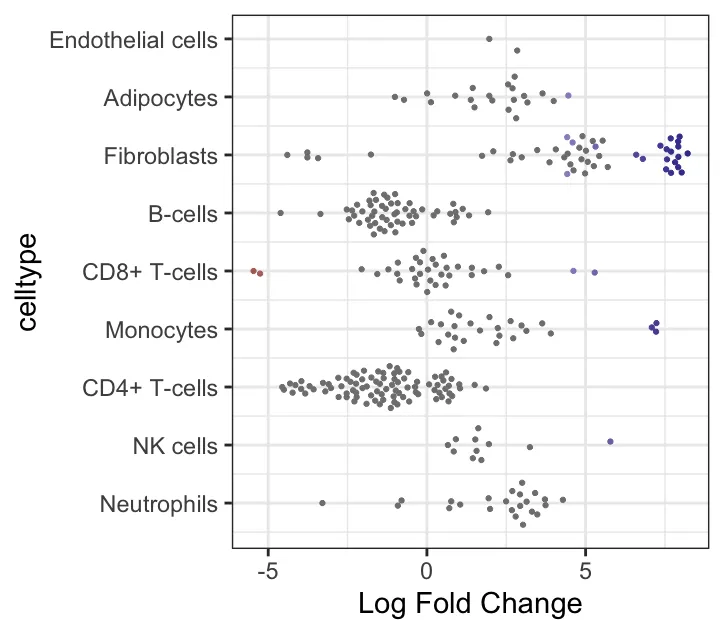
plotDAbeeswarm(da_results, group.by = "celltype",alpha = 0.9) # alpha默认0.1
celltype_fraction 直方图:展示了邻域中某种特定细胞类型的比例分布(celltype_fraction)。Y轴代表邻域的数量,X轴表示邻域中细胞类型所占的比例(范围从0到1)。大部分邻域的细胞类型比例接近1,说明大多数邻域是单一细胞类型主导的。大多数邻域由某一类型的细胞主导(celltype_fraction 为 1)。这意味着邻域中的细胞几乎都是同一种类型,没有明显的混合细胞类型。这种结果通常出现在较为纯净的细胞群或具有显著分离特征的细胞亚群。
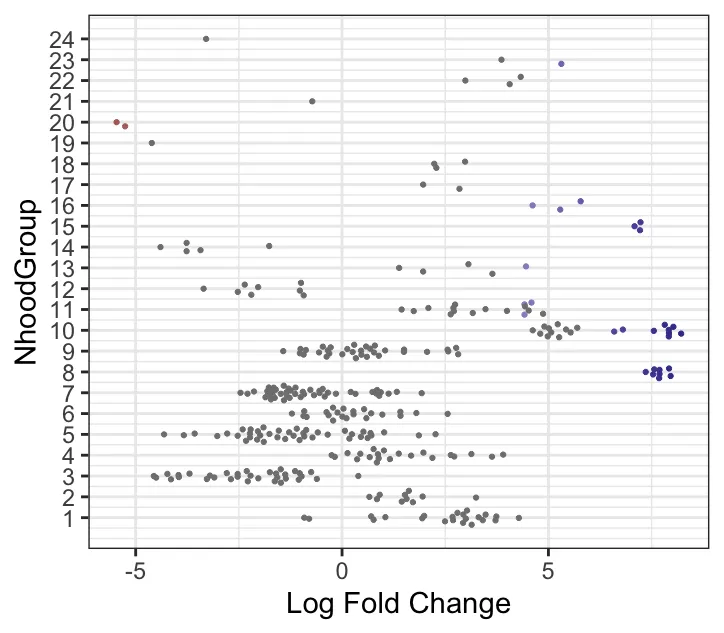
logFC 与细胞类型关联的 beeswarm 图:展示了不同细胞类型的 Log Fold Change(logFC)在不同邻域中的分布。Y轴表示细胞类型,X轴表示logFC(即不同处理组之间在邻域中的差异丰度变化)。每个点代表一个邻域,其中不同的颜色代表不同的显著性水平(根据FDR或p值筛选)。每个细胞类型的点代表其在不同邻域中的表现。图中logFC值分布较为广泛的细胞类型,如Fibroblasts和CD8+ T-cells,在不同处理组间表现出显著的差异丰度变化。而某些细胞类型,如Endothelial cells,可能在各组间差异较小。logFC较大的值(例如正向logFC较大)表明在“treat”组中这些邻域的丰度较高。(笔者的示例数据没有很大差异)
12.找到DA群体的marker
## Add log normalized count to Milo object
scRNA <- logNormCounts(scRNA)
# alpha默认0.1,所以SpatialFDR < 0.9是修改过的
da_results$NhoodGroup <- as.numeric(da_results$SpatialFDR < 0.9 & da_results$logFC < 0)
da_nhood_markers <- findNhoodGroupMarkers(scRNA, da_results,
subset.row = rownames(scRNA),
aggregate.samples = TRUE, # 建议设置为TRUE
sample_col = "orig.ident")
head(da_nhood_markers)
# GeneID logFC_0 adj.P.Val_0 logFC_1 adj.P.Val_1
# 1 AL627309.1 -0.0007221833 0.9413661 0.0007221833 0.9413661
# 2 AL627309.3 -0.0001256128 0.9413661 0.0001256128 0.9413661
# 3 AL645608.1 -0.0156444426 0.2788002 0.0156444426 0.2788002
# 4 AL669831.5 0.0100744381 0.9413661 -0.0100744381 0.9413661
# 5 FAM41C 0.0076732647 0.9413661 -0.0076732647 0.9413661
# 6 FAM87B -0.0056002509 0.3247399 0.0056002509 0.3247399
12.1.自动对邻域进行分组
#在许多情况下,DA邻域位于KNN图的不同区域,将所有重要的 DA 群体分组在一起可能并不理想,因为它们可能包括非常不同细胞类型的细胞。
# 对于这种场景,开发者用了一个邻里函数,它使用社区检测根据(1)两个邻域之间共享的小区数量将邻域划分为组;(2)DA 社区的倍数变化方向;(3)折叠变化的差异。
## Run buildNhoodGraph to store nhood adjacency matrix
scRNA <- buildNhoodGraph(scRNA)
## Find groups
da_results <- groupNhoods(scRNA, da_results,
max.lfc.delta = 2,da.fdr = 0.9)#da.fdr默认0,1
head(da_results)
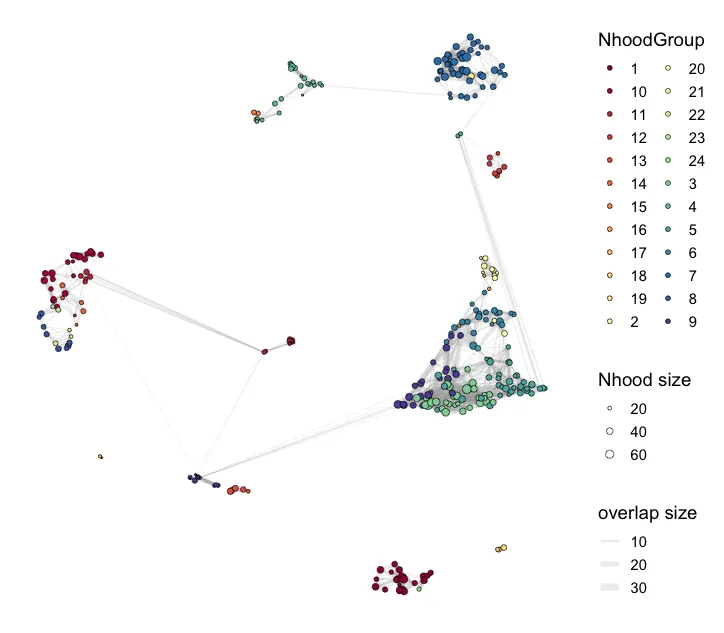
plotNhoodGroups(scRNA, da_results, layout="UMAP")
plotDAbeeswarm(da_results, "NhoodGroup",alpha = 0.9) #alpha默认是0.1
这个步骤是按照DA的结果进行自动划分簇,还可以找到差异基因。max.lfc.delta这个值可以自行修改! 正式分析的时候需要探索。
12.2 找到特征基因
## Exclude zero counts genes
keep.rows <- rowSums(logcounts(scRNA)) != 0
scRNA <- scRNA[keep.rows, ]
## Find HVGs
dec <- modelGeneVar(scRNA)
hvgs <- getTopHVGs(dec, n=2000)
head(hvgs)
# [1] "CFD" "DCN" "MGP" "CXCL8" "MT2A" "S100A9"
# 找到每个邻里群的one-vs-all差异基因表达
nhood_markers <- findNhoodGroupMarkers(scRNA, da_results, subset.row = hvgs,
aggregate.samples = TRUE, sample_col = "orig.ident")
head(nhood_markers)[1:4,1:4]
# GeneID logFC_1 adj.P.Val_1 logFC_2
# 1 A2M -0.43376837 0.35815985 -0.43132559
# 2 AAK1 -0.55965379 0.03462636 0.74914069
# 3 ABCA1 0.20200482 0.46368550 -0.31103802
# 4 ABCA10 -0.08910405 0.50766539 -0.09984058
# 找到group2的差异基因
gr2_markers <- nhood_markers[c("logFC_2", "adj.P.Val_2")]
colnames(gr2_markers) <- c("logFC", "adj.P.Val")
head(gr2_markers[order(gr2_markers$adj.P.Val), ])
# 如果明确了对第二组细胞感兴趣
nhood_markers <- findNhoodGroupMarkers(scRNA, da_results, subset.row = hvgs,
aggregate.samples = TRUE, sample_col = "orig.ident",
subset.groups = c("2")
)
head(nhood_markers)
# logFC_2 adj.P.Val_2 GeneID
# GNLY 4.529267 9.977073e-54 GNLY
# CLIC3 1.273255 4.886386e-40 CLIC3
# PRF1 2.186990 4.886386e-40 PRF1
# KLRF1 1.310507 2.133371e-35 KLRF1
# KLRD1 2.746359 9.425077e-32 KLRD1
# GZMB 2.731049 3.720297e-29 GZMB
# 如果明确了相比较的邻域
nhood_markers <- findNhoodGroupMarkers(scRNA, da_results, subset.row = hvgs,
subset.nhoods = da_results$NhoodGroup %in% c('11','2'),
aggregate.samples = TRUE, sample_col = "orig.ident")
head(nhood_markers)
# GeneID logFC_2 adj.P.Val_2 logFC_11 adj.P.Val_11
# 1 A2M -1.1629210 0.004322766 1.1629210 0.004322766
# 2 AAK1 0.6881532 0.038952082 -0.6881532 0.038952082
# 3 ABCA1 -0.2253056 0.063886117 0.2253056 0.063886117
# 4 ABCA10 -0.1581780 0.064434029 0.1581780 0.064434029
# 5 ABCA6 -0.3517706 0.019652787 0.3517706 0.019652787
# 6 ABCA7 -0.1191482 0.307535525 0.1191482 0.307535525
12.3 差异基因可视化
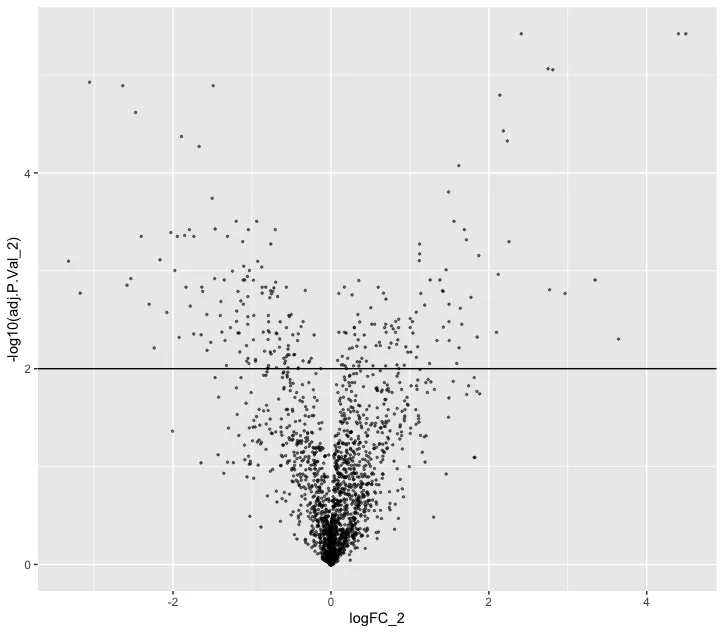
ggplot(nhood_markers, aes(logFC_2,-log10(adj.P.Val_2 ))) +
geom_point(alpha=0.5, size=0.5) +
geom_hline(yintercept = 2)
# 筛选标准可以自行修改
markers <- rownames(nhood_markers)[nhood_markers$adj.P.Val_2 < 0.01 & nhood_markers$logFC_2 > 0]
plotNhoodExpressionGroups(scRNA, da_results, features=markers,
subset.nhoods = da_results$NhoodGroup %in% c('11','2'),
scale=TRUE,
grid.space = "fixed")
12.4 同一邻域但是不同干预的细胞比较
dge_9 <- testDiffExp(scRNA, da_results, design = ~ group,
meta.data = data.frame(colData(scRNA)),
subset.row = rownames(scRNA),
subset.nhoods=da_results$NhoodGroup=="9")
dge_9
# $`9`
# logFC AveExpr t P.Value adj.P.Val B Nhood.Group
# KLHL17 0.017612117 0.011781245 1.4215277 0.1558631 0.6982049 -9.789123 9
# LINC00115 0.026119993 0.041024269 1.1226770 0.2621795 0.6982049 -10.168746 9
# AL627309.1 0.002904512 0.001411100 1.0242142 0.3062900 0.6982049 -10.274403 9
# SAMD11 -0.006652596 0.003305143 -1.0015076 0.3171252 0.6982049 -10.297400 9
# NOC2L -0.030967073 0.180208912 -0.9373189 0.3491025 0.6982049 -10.359634 9
# AL669831.5 -0.005367254 0.007952223 -0.5865251 0.5578198 0.9296997 -10.627143 9
# FAM41C -0.002584961 0.014220774 -0.1830747 0.8548227 1.0000000 -10.782673 9
# AL627309.3 0.000000000 0.000000000 0.0000000 1.0000000 1.0000000 -10.799468 9
# FAM87B 0.000000000 0.000000000 0.0000000 1.0000000 1.0000000 -10.799468 9
# AL645608.1 0.000000000 0.000000000 0.0000000 1.0000000 1.0000000 -10.799468 9
需要提醒一下,其中很多参数笔者做了修改也做了注释,使用的时候需要先从默认值开始分析!
参考资料:
-
Differential abundance testing on single-cell data using k-nearest neighbor graphs. Nat Biotechnol. 2022 Feb;40(2):245-253.
-
miloR:https://rawcdn.githack.com/MarioniLab/miloR/7c7f906b94a73e62e36e095ddb3e3567b414144e/vignettes/milo_gastrulation.html#5_Finding_markers_of_DA_populations
-
单细胞天地:https://mp.weixin.qq.com/s/yYTxb_kOjeRSkzzuUoI4mA
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -















 860
860

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









