







简 介
当前用于比较分析单细胞数据集的计算工作流程在测试不同实验条件下的差异丰度时,通常使用离散 cluster 作为输入。然而,聚类并不总是提供适当的分辨率,也不能捕获连续的轨迹。在这里,我们提出了Milo,一个可扩展的统计框架,通过在k近邻图上将单细胞分配到部分重叠的邻域来执行差异丰度测试。使用模拟和单细胞RNA测序(scRNA-seq)数据,表明 Milo 可以识别因离散细胞成 cluster 而被掩盖的扰动,它在批效应中保持错误发现率控制,并且它优于其他差异丰度测试策略。Milo 确定了衰老小鼠胸腺中命运偏向性上皮前体的衰退,并确定了人类肝硬化中多个谱系的扰动。由于Milo是基于细胞-细胞相似性结构,它可能也适用于scRNA-seq以外的单细胞数据。

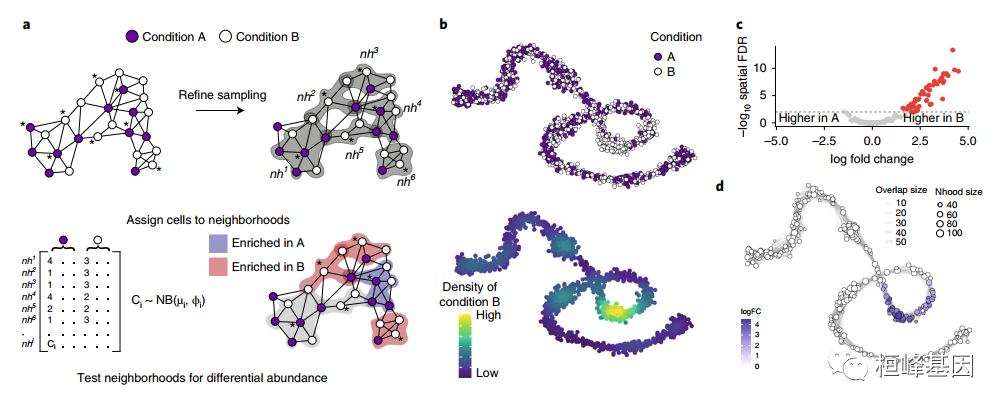
作为差分富度图邻域的扰动细胞状态检测。a,Milo工作流程示意图。邻域定义在索引细胞上,使用图采样算法选择。根据实验设计对细胞进行定量,生成计数表。每个社区细胞计数使用负二项GLM建模,并进行假设检验以确定差异丰富的社区。b, KNN图的力定向布局,表示从两个实验条件下采样的细胞的模拟连续轨迹(上图:条件A,紫色;条件B,白色;下图:条件B中细胞的核密度。c,使用Milo进行假设检验,准确且特异性地检测到差异丰富的邻域(FDR为1%)。红点表示不同数量的邻域。d,米洛差分丰度检验结果的图示。节点是邻域,通过它们的对数折叠变化来着色。非差异丰富的邻域(FDR 1%)是白色的,大小对应于邻域中的细胞数量。图边描述了相邻邻域之间共享的单细胞数量。节点的布局由邻域索引单细胞在单个细胞的力定向嵌入中的位置决定。
软件包安装
安装较为简单,直接安装即可:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("miloR")
## Install development version
devtools::install_github("MarioniLab/miloR", ref="devel")数据读取
读入一个典型的单细胞数据,包括meta和表达矩阵,这里的矩阵是 SingleCellExperiment:
library(miloR)## 载入需要的程辑包:edgeR## 载入需要的程辑包:limmadata(sim_trajectory)
milo.meta <- sim_trajectory$meta
head(milo.meta)## cell_id group_id Condition Replicate Sample
## 1 C1 M2 B R1 B_R1
## 2 C2 M2 B R1 B_R1
## 3 C3 M1 A R1 A_R1
## 4 C4 M2 B R1 B_R1
## 5 C5 M1 B R1 B_R1
## 6 C6 M1 B R1 B_R1milo.obj <- Milo(sim_trajectory$SCE)
milo.obj## class: Milo
## dim: 500 500
## metadata(0):
## assays(2): counts logcounts
## rownames(500): G1 G2 ... G499 G500
## rowData names(0):
## colnames(500): C1 C2 ... C499 C500
## colData names(2): cell_id group_id
## reducedDimNames(1): PCA
## mainExpName: NULL
## altExpNames(0):
## nhoods dimensions(2): 1 1
## nhoodCounts dimensions(2): 1 1
## nhoodDistances dimension(1): 0
## graph names(0):
## nhoodIndex names(1): 0
## nhoodExpression dimension(2): 1 1
## nhoodReducedDim names(0):
## nhoodGraph names(0):
## nhoodAdjacency dimension(2): 1 1实例操作
差异分析
对workflow的基本描述:
1,数据预处理与降维
2,minimizing batch effects.如harmony等整合多sample的算法,但不要使用conos
3,building the KNN graph
4,definition of cell neighbourhoods and index cell samplling
5,testing for differencial abundance in neighbourhoods
milo.obj <- buildGraph(milo.obj, k=20, d=30)## Constructing kNN graph with k:20milo.obj <- makeNhoods(milo.obj, k=20, d=30, refined=TRUE, prop=0.2)## Checking valid object## Running refined sampling with reduced_dimmilo.obj <- calcNhoodDistance(milo.obj, d=30)## as(<dgTMatrix>, "dgCMatrix") is deprecated since Matrix 1.5-0; do as(., "CsparseMatrix") insteadmilo.obj <- countCells(milo.obj, samples="Sample", meta.data=milo.meta)## Checking meta.data validity## Counting cells in neighbourhoodsmilo.design <- as.data.frame(xtabs(~ Condition + Sample, data=milo.meta))
milo.design <- milo.design[milo.design$Freq > 0, ]
rownames(milo.design) <- milo.design$Sample
milo.design <- milo.design[colnames(nhoodCounts(milo.obj)),]
milo.res <- testNhoods(milo.obj, design=~Condition, design.df=milo.design)## Using TMM normalisation## Performing spatial FDR correction with k-distance weightinghead(milo.res)## logFC logCPM F PValue FDR Nhood SpatialFDR
## 1 -1.2323695 15.20142 14.77221137 0.001157153 0.003085743 1 0.003009444
## 2 -0.2608166 15.18022 0.53107695 0.506271278 0.558644169 2 0.560950918
## 3 0.1058813 14.92559 0.07601276 0.812617751 0.813801465 3 0.813801465
## 4 -0.4161003 15.51904 1.75022259 0.220752573 0.261632679 4 0.259524289
## 5 0.5080137 15.50203 2.29454278 0.164196289 0.202087741 5 0.199463470
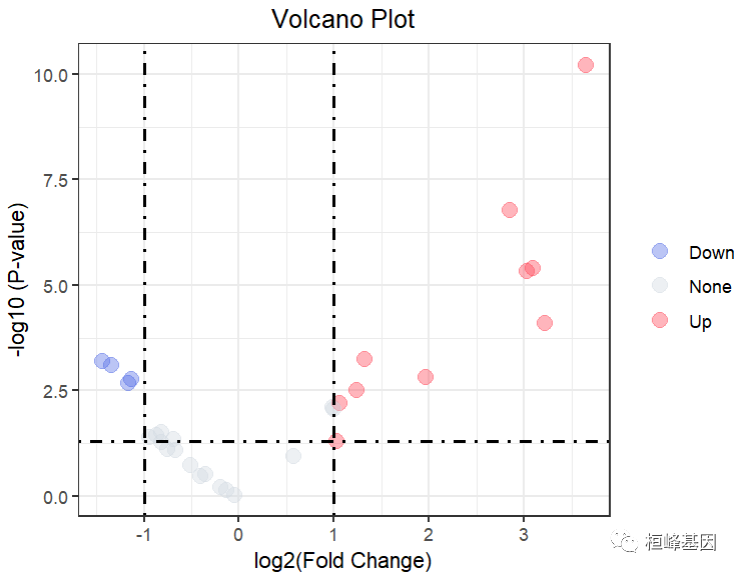
## 6 0.6824722 15.18011 3.12914902 0.106498870 0.136318554 6 0.135804176绘制火山图
首先设置pvalue和logFC的阈值,然后根据阈值分别为上调基因设置‘up’,下调基因设置‘Down’,无差异设置‘None’,保存到change列。这里的change列用来设置火山图点的颜色
cut_off_pvalue = 0.05
cut_off_logFC = 1
#
milo.res$Sig = ifelse(milo.res$PValue < cut_off_pvalue &
abs(milo.res$logFC) >= cut_off_logFC,
ifelse(milo.res$logFC> cut_off_logFC ,'Up','Down'),'None')
table(milo.res$Sig)##
## Down None Up
## 4 19 9library(ggplot2)## Warning: 程辑包'ggplot2'是用R版本4.2.3 来建造的ggplot(milo.res, aes(x = logFC, y = -log10(PValue), colour=Sig)) +
geom_point(alpha=0.4, size=3.5) +
scale_color_manual(values=c("#546de5", "#d2dae2","#ff4757"))+
# 辅助线
geom_vline(xintercept=c(-1,1),lty=4,col="black",lwd=0.8) +
geom_hline(yintercept = -log10(cut_off_pvalue),
lty=4,col="black",lwd=0.8) +
# 坐标轴
labs(x="log2(Fold Change)",
y="-log10 (P-value)")+
theme_bw()+
ggtitle("Volcano Plot")+
# 图例
theme(plot.title = element_text(hjust = 0.5),
legend.position="right",
legend.title = element_blank()
)
Reference
Dann, E., Henderson, N.C., Teichmann, S.A. et al. Differential abundance testing on single-cell data using k-nearest neighbor graphs. Nat Biotechnol (2021).
单细胞生信分析教程
桓峰基因公众号推出单细胞生信分析教程并配有视频在线教程,目前整理出来的相关教程目录如下:
SCS【4】单细胞转录组数据可视化分析 (Seurat 4.0)
SCS【6】单细胞转录组之细胞类型自动注释 (SingleR)
SCS【7】单细胞转录组之轨迹分析 (Monocle 3) 聚类、分类和计数细胞
SCS【8】单细胞转录组之筛选标记基因 (Monocle 3)
SCS【9】单细胞转录组之构建细胞轨迹 (Monocle 3)
SCS【10】单细胞转录组之差异表达分析 (Monocle 3)
SCS【11】单细胞ATAC-seq 可视化分析 (Cicero)
SCS【12】单细胞转录组之评估不同单细胞亚群的分化潜能 (Cytotrace)
SCS【13】单细胞转录组之识别细胞对“基因集”的响应 (AUCell)
SCS【15】细胞交互:受体-配体及其相互作用的细胞通讯数据库 (CellPhoneDB)
SCS【16】从肿瘤单细胞RNA-Seq数据中推断拷贝数变化 (inferCNV)
SCS【17】从单细胞转录组推断肿瘤的CNV和亚克隆 (copyKAT)
SCS【18】细胞交互:受体-配体及其相互作用的细胞通讯数据库 (iTALK)
SCS【21】单细胞空间转录组可视化 (Seurat V5)
SCS【22】单细胞转录组之 RNA 速度估计 (Velocyto.R)
SCS【24】单细胞数据量化代谢的计算方法 (scMetabolism)
SCS【25】单细胞细胞间通信第一部分细胞通讯可视化(CellChat)
SCS【26】单细胞细胞间通信第二部分通信网络的系统分析(CellChat)
SCS【28】单细胞转录组加权基因共表达网络分析(hdWGCNA)
SCS【29】单细胞基因富集分析 (singleseqgset)
SCS【30】单细胞空间转录组学数据库(STOmics DB)
SCS【31】减少障碍,加速单细胞研究数据库(Single Cell PORTAL)
SCS【32】基于scRNA-seq数据中推断单细胞的eQTLs (eQTLsingle)
SCS【33】单细胞转录之全自动超快速的细胞类型鉴定 (ScType)
SCS【34】单细胞/T细胞/抗体免疫库数据分析(immunarch)
SCS【35】单细胞转录组之去除双细胞 (DoubletFinder)
利用这个软件包实现了快速计算单细胞差异丰度,有需求的老师可以联系桓峰基因,关注桓峰基因公众号,轻松学生信,高效发文章!
号外号外,桓峰基因单细胞生信分析免费培训课程即将开始,快来报名吧!
桓峰基因,铸造成功的您!
未来桓峰基因公众号将不间断的推出单细胞系列生信分析教程,
敬请期待!!
桓峰基因官网正式上线,请大家多多关注,还有很多不足之处,大家多多指正!
http://www.kyohogene.com/
桓峰基因和投必得合作,文章润色优惠85折,需要文章润色的老师可以直接到网站输入领取桓峰基因专属优惠券码:KYOHOGENE,然后上传,付款时选择桓峰基因优惠券即可享受85折优惠哦!https://www.topeditsci.com/


















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









