







前言:因为有人私下来问我,我才发现有人可能不知道MGLTools其实就是Autodock Tools,有的人也会称它为ADT,因为是通过adt.bat打开的。本文全部用ADT指代。
同时本来也不准备发这篇笔记的,但是后台有一些粉丝才问,才总结。
1.使用ADT分析相互作用
导入vina结果的pqbqt文件
Analyze->Dockings->Open Autodock vina result导入MOLxxx_out.pdbqt。选择Single xxx
出现warning弹窗直接选择确定即可,显示如下:
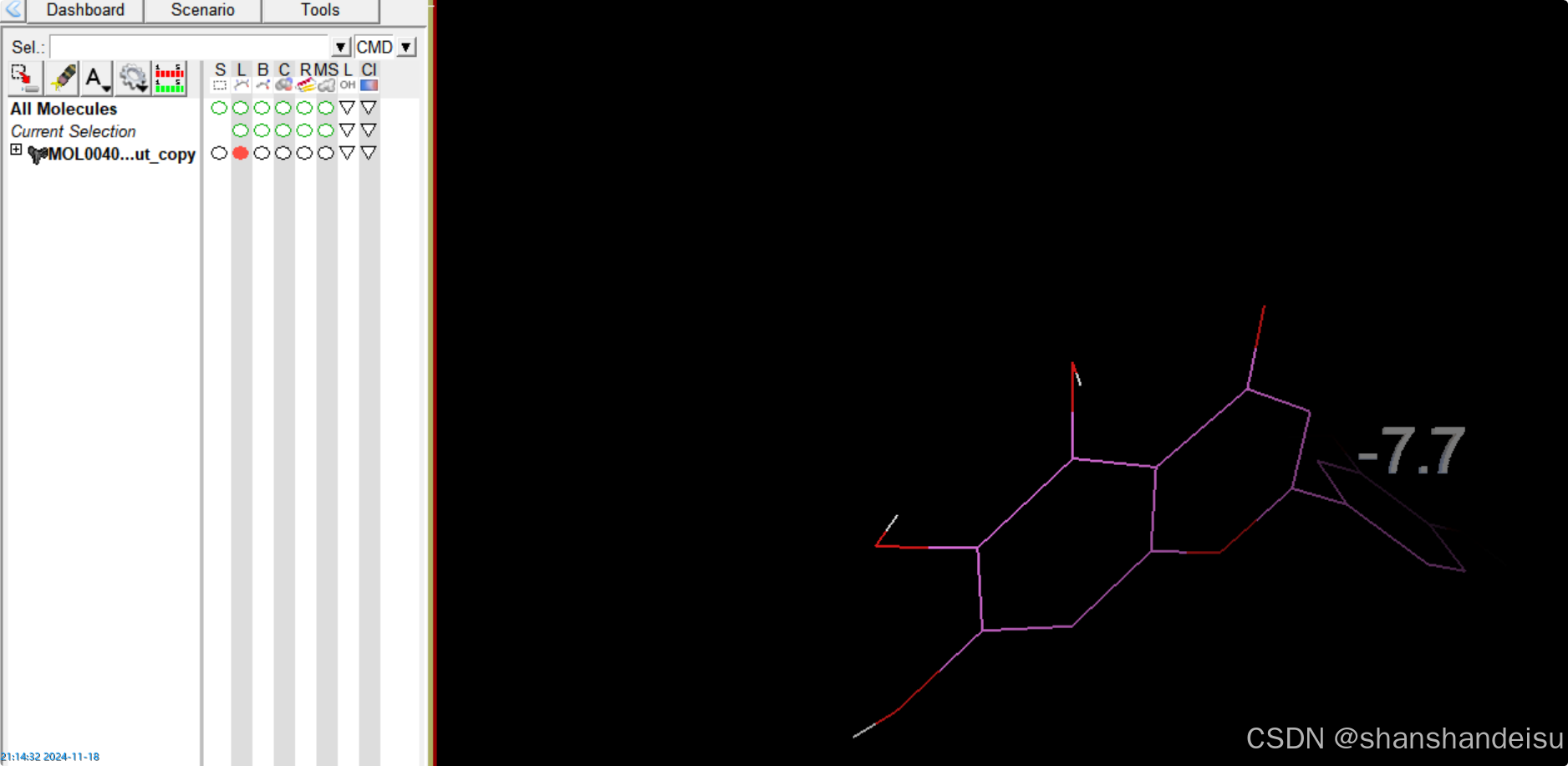
使用键盘方向键←→可以切换构象。每个构象上都可以看到结合能。
导入蛋白并分析
Analyze->Macromolecule->Open导入蛋白的pdbqt文件。
Analyze->Dockings->Show Interactions,便会弹出面板如下:
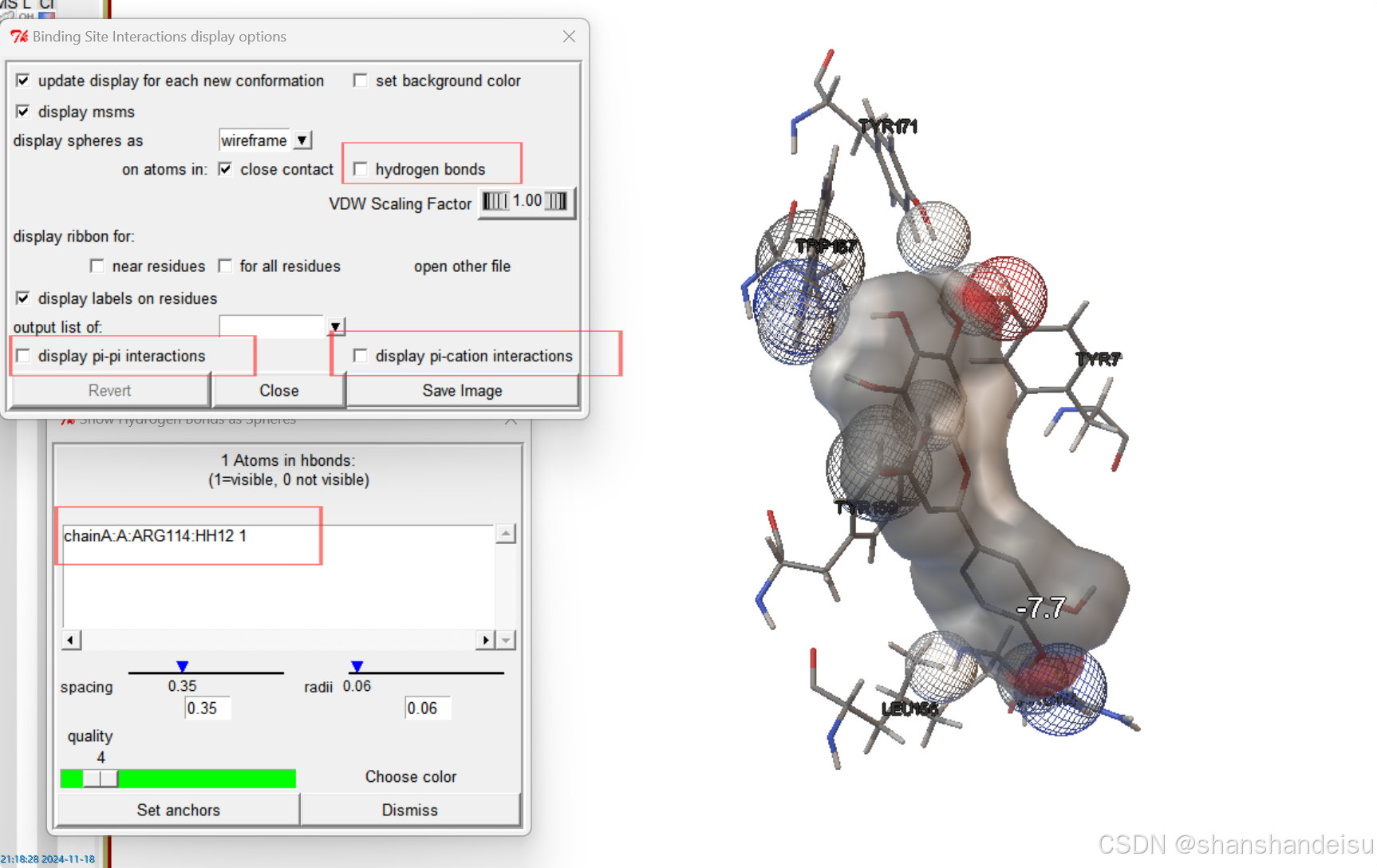
注意画红框的部分,接下来会详细分析。
氢键分析
选中氢键分析,可以发现一些地方从实心球体变成线框。这些变化的部分就是参与了氢键的原子。
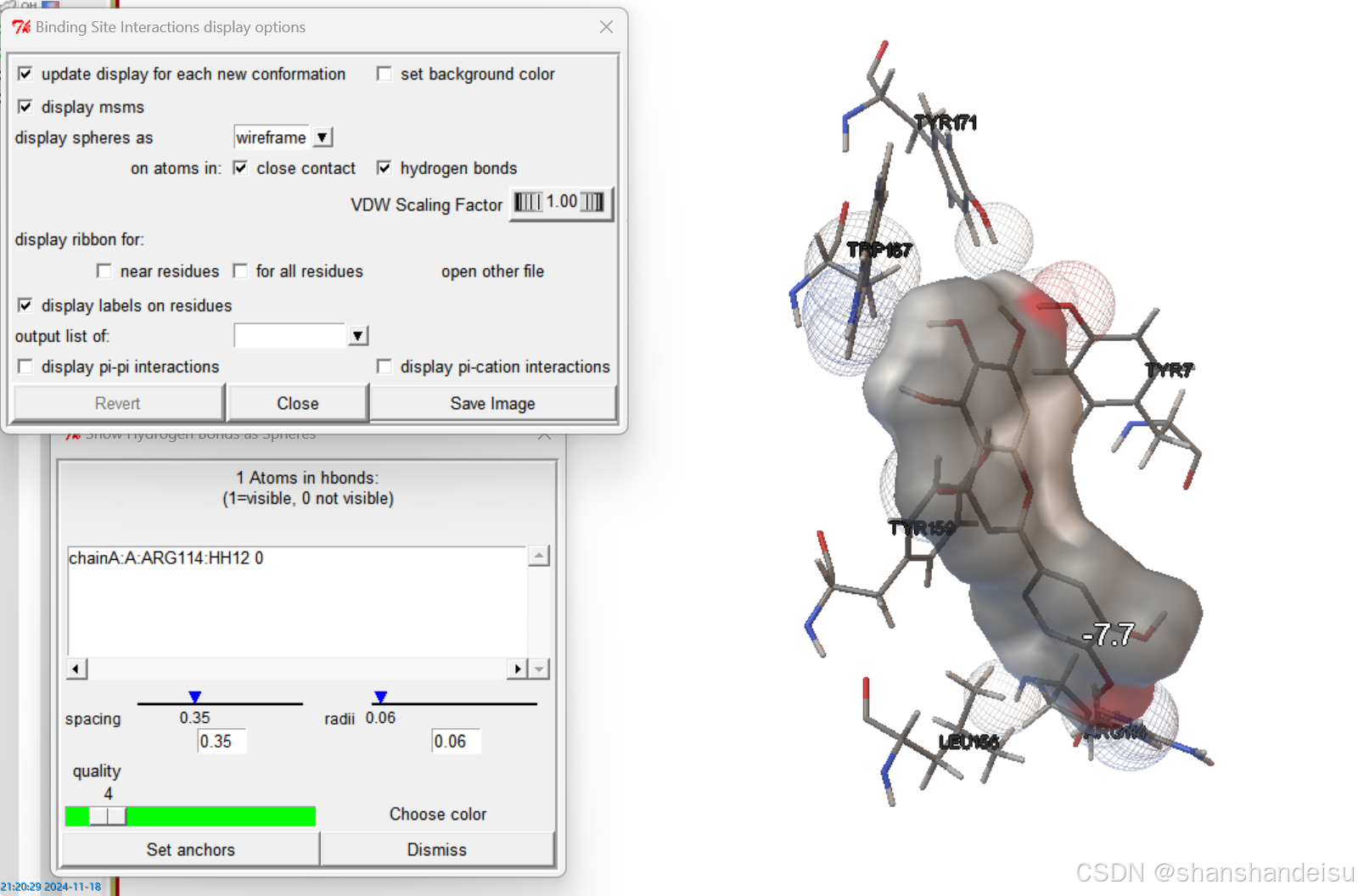
同时,在下面Show Hydrogen Bonds as Spheres面板中显示的chainA:A:ARG114:HH12 0信息,点击后会变为chainA:A:ARG114:HH12 1意思是:
ARG114: 表示这个氢键涉及的氨基酸残基是精氨酸Arginine,其序列位置为114。HH12: 表示这个氢键涉及的氢原子是精氨酸残基Arg114上的第12个氢原子。1: 表示这个氢键的长度为1埃(Å)
pi-pi键分析和pi-cation键分析
选中pi-pi键,显示如下:
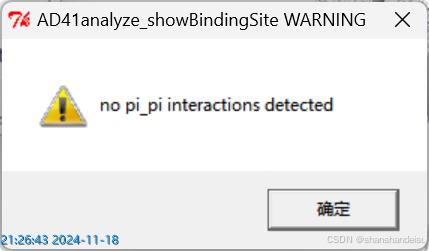
选中pi-cation键,显示如下:
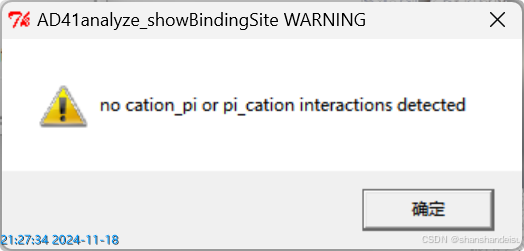
说明没有pi-pi键或pi-cation键或cation-pi键。
2.使用ADT保存配体构象(得到pdb)
在步骤1后,将受体蛋白删除后,file/save/write pdb保存配体的构象。譬如这里命名为MOLxxx_out_1.pdb。
3.使用pymol合并蛋白和配体构象(得到pdb)
将刚刚的lig1.pdb和受体蛋白.pdb(去水去配体处理后的,我这里是chainA.pdb)拖拽进pymol,然后export molecule直接保存为complex.pdb
注意此时不要退出pymol,直接将蛋白和配体隐藏掉后,再拖拽complex.pdb进来,看是否真的是结合好了的蛋白和配体,如下:

再隐藏complex.pdb,查看蛋白和配体。对比看下和刚刚的complex.pdb是否位置一样。
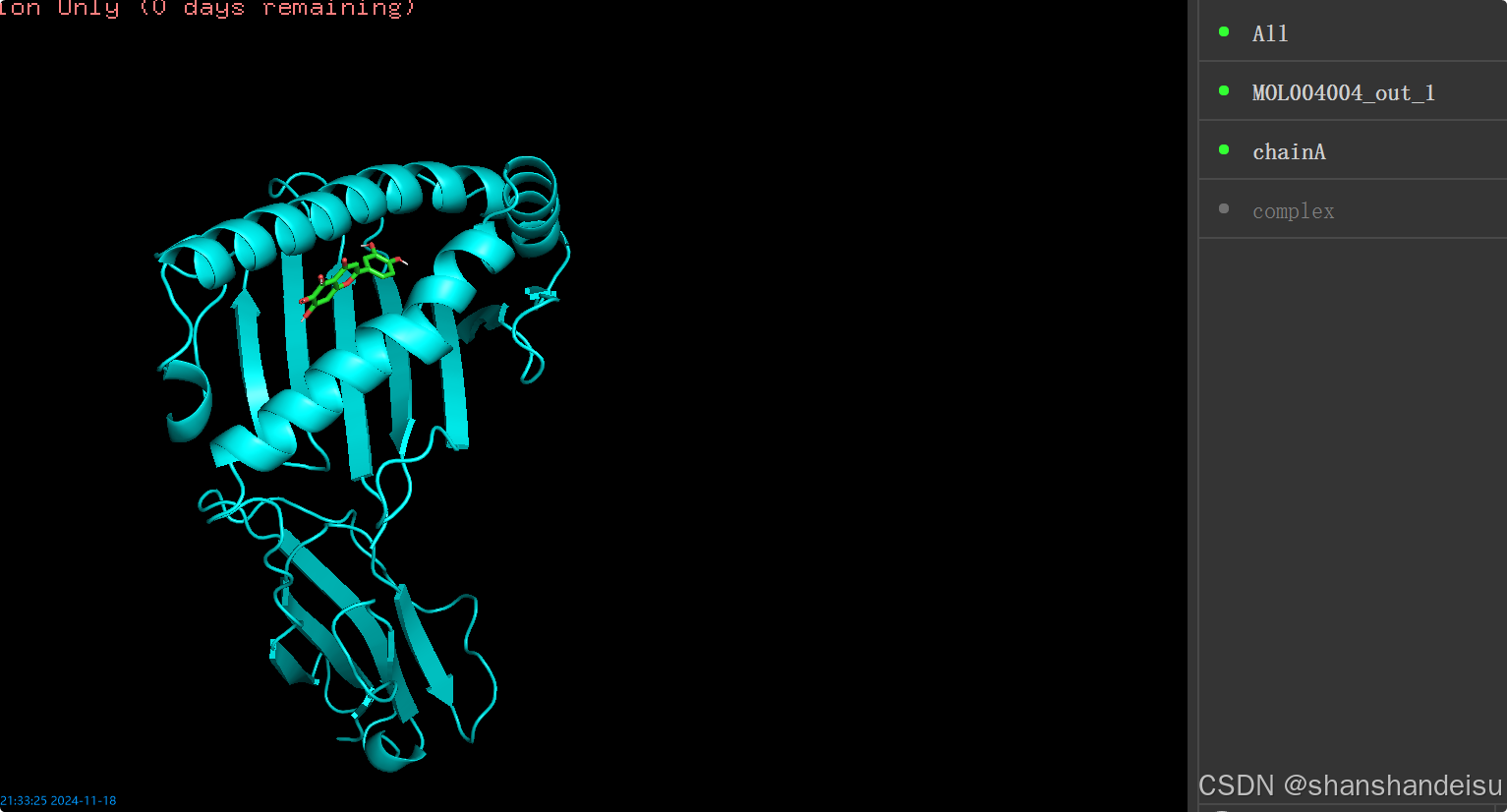
可以发现是一样的哈。
4.使用pymol直接保存配体构象(步骤2的替代)
有时候可能不需要通过ADT来分析相互作用,譬如我就直接选结合能最低的那个构象就好了,那么我们可以直接用pymol来保存配体构象即可。
将vina对接结果的pdbqt拖拽进来,显示如下:
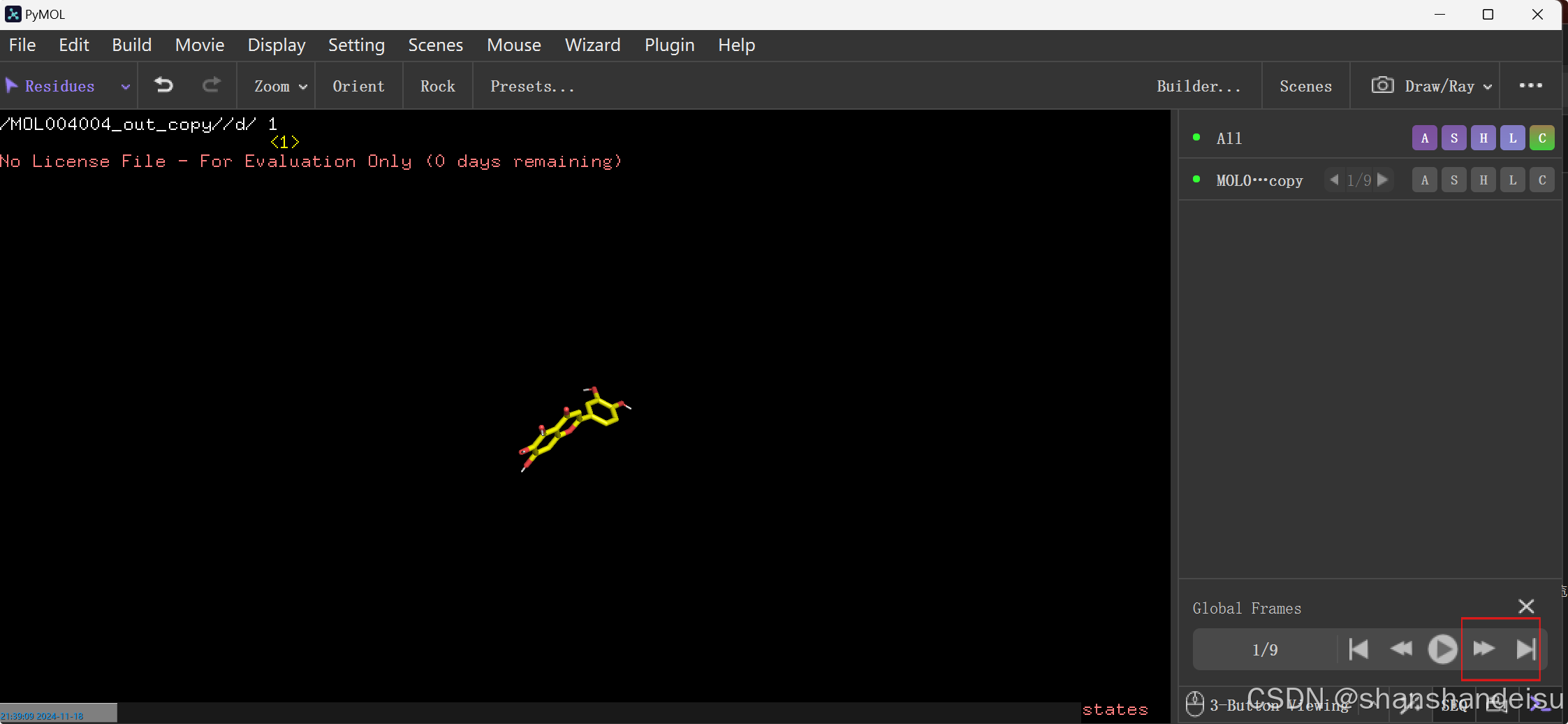
1/9代表着这是第一个构象,因为构象都是对接分数由高到低(结合能由小到大排列)的,所以一般第一个构象就是结合能最低的那个。
然后我们export molecule直接保存为lig1.pdb即可保存配体构象。
当然,点击▶播放键也可以查看9个构象的变化动画,点击⏩的话可以切换到下一个构象,点击尾页的键就是到第九个构象(这个emoji不知道怎么打hhh)。
5.使用pymol直接生成复合物(步骤3的替代)
将vina对接结果的pdbqt拖拽进来
并把受体蛋白.pdb(去水去配体处理后的,我这里是chainA.pdb)拖拽进pymol
然后export molecule直接保存为complex.pdb
也会直接生成第一个配体构象和蛋白结合的复合物体系
不需要像步骤3一样将配体构象lig1.pdb再拖拽进来了。
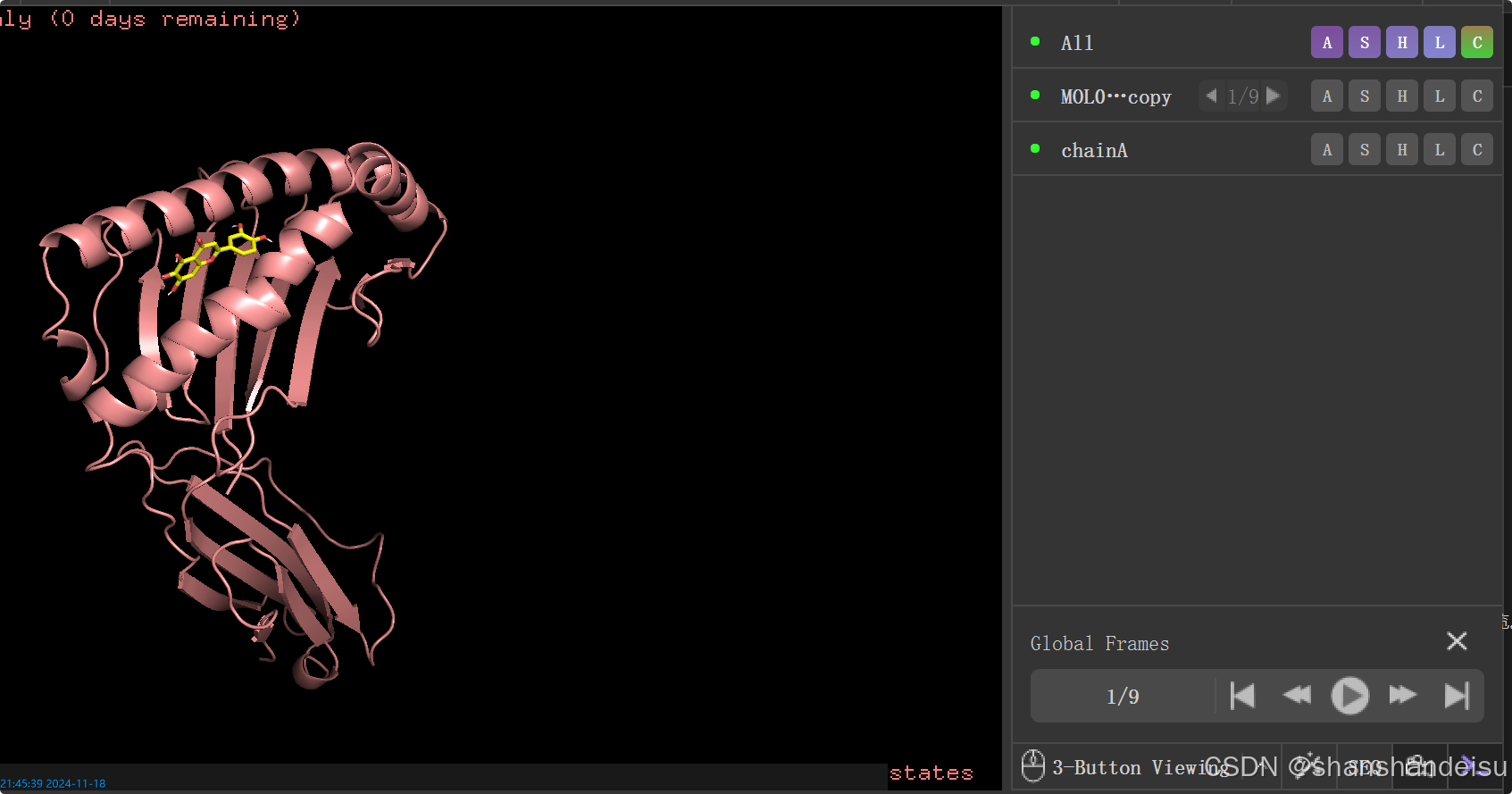
最终生成有9个构象,但是pymol打开pdbqt文件,里面只有一个构象,右下角state显示1/1该如何解决?
解决方案来自b站评论:
在使用vina命令行生成分子对接结果时,存在默认参数–energy_range arg (=3) 表示默认与最优模型能量相差允许为3以内,尝试把3改成5则会显示更多的构象。
6.UCSF Chimera相关操作(不常见)
奇美拉软件也可以进行配体构象的保存以及复合物的生成操作,因为本人不用奇美拉软件,所以感兴趣的同学可以看这个视频:https://www.bilibili.com/video/BV1tf4y1y7TG/
7.vina_split脚本保存配体构象(得到pdbqt)
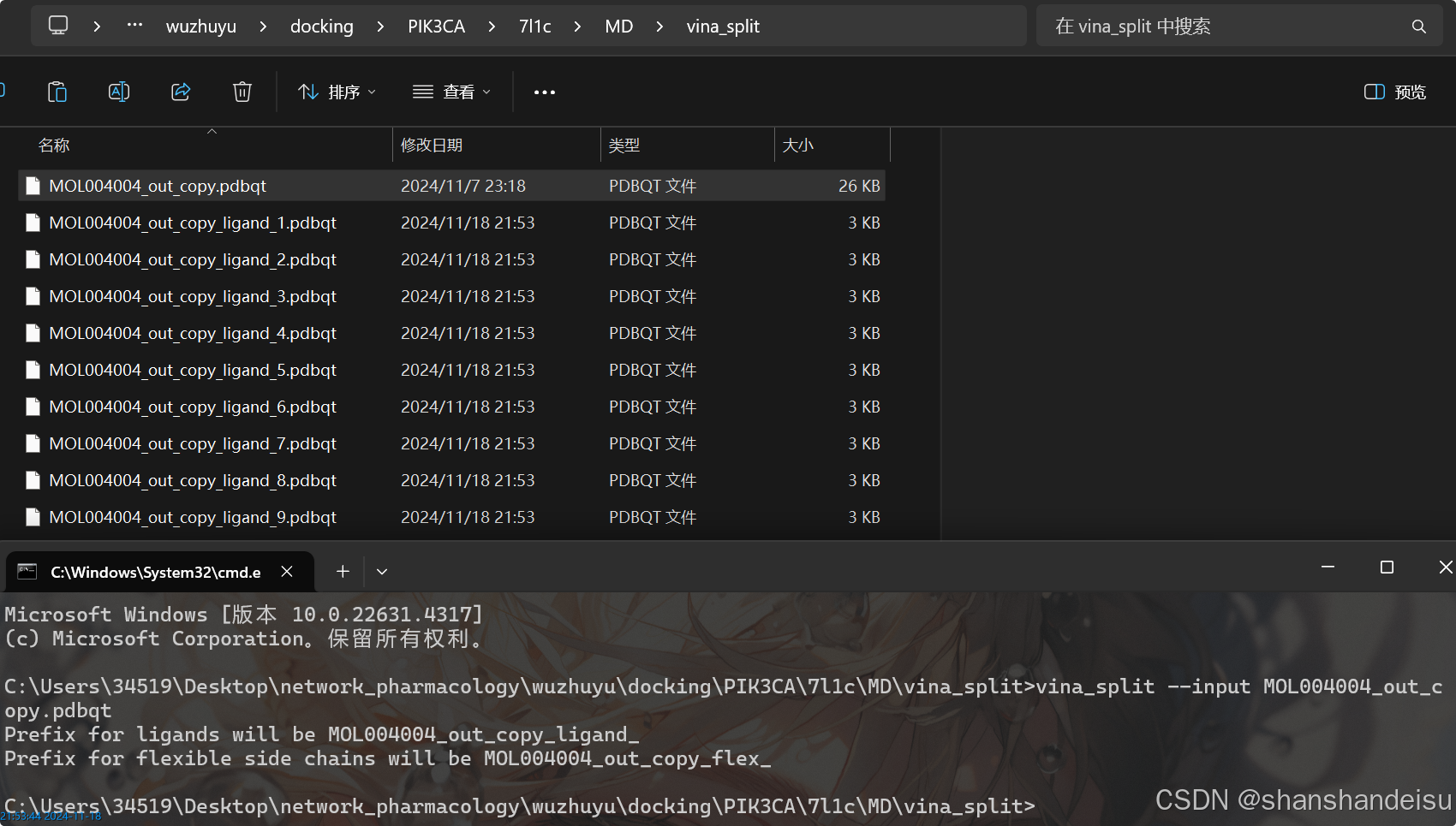
下载autodock vina时,会自动下载工具vina_split脚本,来分离配体构象。
我们准备一个工作目录为vina_split,目录下只存在我们的vina对接pdbqt结果。
然后在该工作目录上打开命令行工具,输入命令:
vina_split --input 你的vina对接pdbqt结果
然后看到输出
prefix for ligands xxx
prefix for flexible xxx
则说明分离成功。
目录下会自动生成分离后的9个构象,每个构想都是原文件名加_ligand_序号,如下:
8.文本操作合并蛋白和配体构象(步骤3的替代,其实不推荐hhh)
在b站上查看分子动力学模拟的教程的话,可以看到很多都是用文本操作来合并蛋白和配体构象的,在这简单演示一下。
注意!合并蛋白和配体构象的话,最好使用同一种格式,要么都是
pdb,要么都是pdbqt。
否则最后pymol检验打开的话,可能会看到一些球型结构显示的原子,这些就是pymol识别不出来的非键原子。
不过因为不同的软件对于pdb和pdbqt,以及pdbqt和pdb格式混合在一起的识别不一样,譬如奇美拉打开可能就不会有球星结构显示的原子。
总之使用同一种格式的话出错的概率会小很多。
记事本打开步骤2生成的配体构象lig1.pdb
记事本打开蛋白chainA.pdb(注意是去水去配体等前置处理后的)
新建一个文本文件complex.txt,将chainA.pdb的记事本内容全部复制进去
然后在最后一行END前,将lig1.pdb的记事本内容插入进去。
最后将complex.txt重命名为complex.pdb,拖拽进pymol检验。可以发现也是成功显示了蛋白配体复合物体系。



















 2686
2686

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











