






根据上一篇文章的选择,关联分析全程,理解+过程,1.5版-CSDN博客
1,如何看QQ和曼哈顿的图
GWAS可视化1 | QQ图和曼哈顿图是嘛意思?_gwas做出来的图叫什么-CSDN博客
如何理解GWAS中Manhattan plot和QQ plot所传递的信息 - 知乎 (zhihu.com)
1.1曼哈顿图
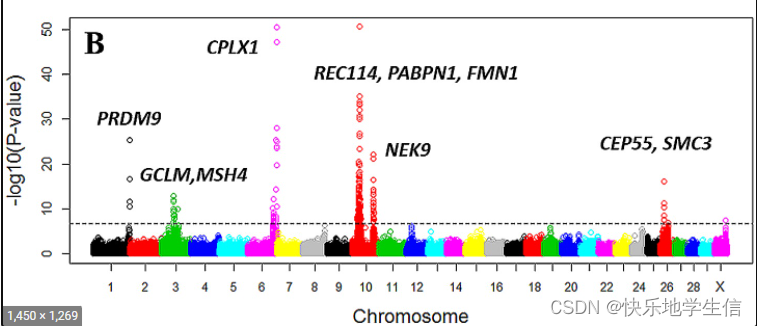
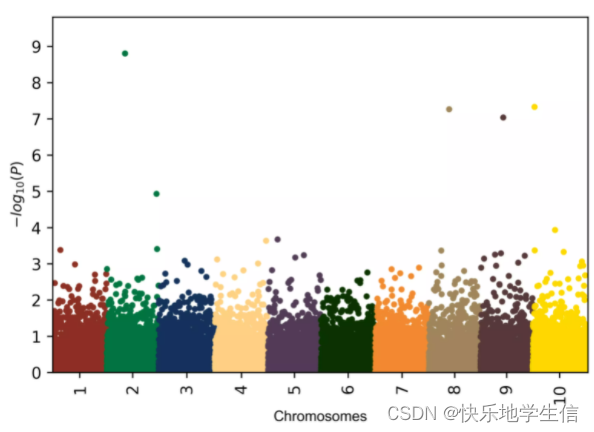
曼哈顿图,Manhattan plot,它是把GWAS分析之后所有SNP位点的p-value在整个基因组上从左到右依次画出来。为了可以更加直观地表达结果,通常都会将p-value转换为-log10(p-value)。这样的话,基因位点-log10(p-value)在Y轴的高度就对应了与表型性状或者疾病的关联程度,关联度越强(即,p-value越低)就越高。而GWAS研究中,一般p-value阈值一般要在10^-6次方甚至10^-8次方以下,也就说曼哈顿图中Y轴大于6甚至大于8的那些SNP位点才是比较值得研究的。
好的曼哈顿图:
GWAS分析中,原理就是SNP位点和控制性状的基因存在LD状态,即SNP的分型可以代表基因的不同分型,所以,真是的显著位点应该是在基因两侧分布的,有一个上升和下降的趋势,比如这样的图:
坏的曼哈顿图:
只有孤零零的一个点,周围没有显著的点,这很有可能是假阳性,因为GWAS分析和培育品种是类似的:一出出一窝!
原文链接:https://blog.csdn.net/yijiaobani/article/details/123507391
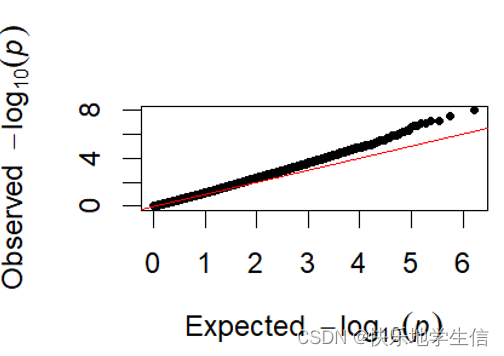
1.2QQ图
QQ plot全称是quantile-quantile plot,也就是分位图,是一种通过比较两个概率分布的分位数从而实现对两个概率分布进行比较的概率图方法(在统计学上较常用)。首先QQ图的x坐标是均匀分布的值(理论值),经过-log10转换了。QQ图的y坐标实际的P值(观测值),经过-log10转换了。
什么意思?
假设我们想要比较某个样本数据的分布与标准正态分布的一致性。我们首先按照从小到大的顺序对样本数据进行排序,然后计算每个数据点的累积概率分布,并将这些累积概率值转换为对应的标准正态分布的分位数。接着,我们将这些标准化后的分位数与标准正态分布的理论分位数进行比较,结果会以散点图的形式呈现在QQ图上。
如果观测数据的分布与标准正态分布一致,那么QQ图上的点将大致落在一条45度直线上。如果观测数据的分布存在偏差,那么QQ图上的点将偏离45度直线,这表明观测数据的分布与标准正态分布存在差异。
根据两篇文章的介绍,
我们认为,P值达到显著性,那就说明不同的SNP分型,对表型数据是有显著性影响的,但是在GWAS中,我们将显著的原因分为两个部分:
第一个部分,是由于突变导致的表型变异,比如AA变异了AT或者TT,导致产奶量增加,AA比如是10Kg,AT是15Kg,TT是20Kg,而且达到极显著水平,我们可以认为该突变导致产奶量变化,使我们关注的位点或者基因。GWAS的目的就是找到这样的位点,进而找到这样的基因。
第二个部分,是随机遗传漂变,它显示的统计显著性只是偶然,并不是该位点真的影响表型值。随机漂变,是随机在染色体上变化,它符合均匀分布,所以一定范围内有一定的概率。这些位点,使我们需要排除的。
如果所谓的变异都是遗传漂变引起的:
那就是这样一个图:横坐标和纵坐标都是在一条直线上,他们是完全一致的,因为X坐标是模拟的均匀分布的P值,而Y坐标是真是的P值,它与X一致,就说明它的分布也是均匀分布的,那我们就可以认为它是随机漂变的产物。
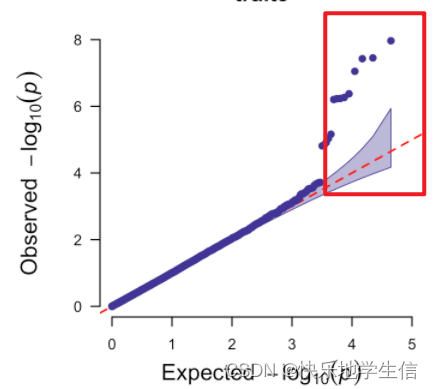
如果所谓的变异都是遗传漂变引起+真实变异引起的:
那它的图应该是这样的:刚开始的位点,比如-log10在3之前,都是和均匀分布是一致的,是随机漂变导致的。在大于3之后,均匀分布还是在直线上,但是由于随机漂变+真实位点的存在,开始偏离直线,并且上翘,这些上翘的位点就是我们关注的位点。所以,好的GWAS分析,有结果的QQ图,都是前期在直线上,后面上翘。有点翘的QQ图才是好的QQ图。
而且,我们知道基因组上的随机漂变是一定存在的,这就表现在QQ-plot的前半部分里。比较好的结果是,当p-value < 10^-3时,GWAS结果开始与均匀分布出现快速分离——也就是说,自然选择的力量明显地显示出来了,使得结果在群体中快速摆脱随机性,最后看到一个高高翘起的QQ-plot。这时基本就可以断定,我们所研究的表型和基因型之间是存在着显著相关的自然选择作用的。
原文链接:https://blog.csdn.net/yijiaobani/article/details/123507391

2.QQ图和曼哈顿图的可视化代码
2,1我们QQ和曼哈顿的图在R里面有自带的包,
使用QQ和曼哈顿图直接画
setwd("lunjin")#切到自己的路径
install.packages("qqman")#安装library("qqman")
vignette('qqman') #可以查看,RStudio里面qqman和manhattan自带的help,说的很详细,里面有自带的文件数据应该如何处理的格式,非常方便
results_assoc <- read.table("wenjain", header = T)#先QQ判断性状关联是否被基因组控制
z = qnorm(results_assoc$P/2) #将p值的一半转为正态分布数
lambda = round(median(z^2, na.rm = T)/0.454, 3) #计算lambda值,即基因组膨胀系数,λ越偏离1,就表明存在群体分层的现象越严重,容易有假阳性结果
qq(results_assoc$P, main = "Q-Q plot of GWAS p-values",
xlim = c(0, 7), ylim = c(0,8),
pch = 18, #pch参数用于设置散点的形状
col = "blue4",
cex = 0.5, #cex参数用于设置散点的大小
las = 1)#las参数用于设置标签的方向#当时这两种代码都能跑
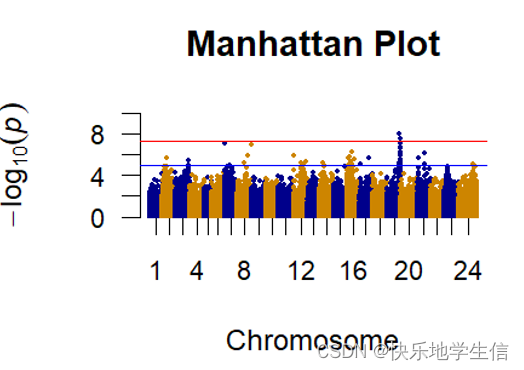
manhattan(results_assoc, main = "Manhattan Plot", ylim = c(0, 10), cex = 0.6,
cex.axis = 0.9,
col = c("blue4", "orange3"),
suggestiveline = -log10(1e-05),
genomewideline = -log10(5e-08))manhattan(results_assoc, main = "Manhattan Plot",
ylim = c(0,10), cex = 0.6, #reduce the point size to 60%点
cex.axis = 0.9,# reduce the font size of the axis labels to 90%字
col = c("blue4", "orange3"),
annotatePval = NULL,
annotateTop = T)
全部流程大概是这样的了,在把图片导出来就可以了。(注意参数可以自己调)
2.2ggpolt画图
ggplot2画图,注意的是+直接加图册画, 和用procreate添加图层一样,参数自己去调查,这里就不多说了。
library(ggplot2)
p <- ggplot(Snp_pos, aes(x=Chromosome, y=-log10(P))) +
geom_point(aes(color = as.factor(CHR)), alpha = 0.8, size = 0.6) +
scale_color_manual(values = rep(c("blue4", "orange3"), 24)) +
scale_x_continuous(label = X_axis$CHR, breaks= X_axis$center ) +
scale_y_continuous(limits = c(0, 10), expand = c(0,0))+
geom_hline(yintercept = c(-log10(1e-05), -log10(5e-08)), color = c('#003399', 'red'), linetype = c("twodash", "solid")) +
labs(x = "chromosome", y = "-log10(P)") +
theme_bw() +
theme(
legend.position="none",
panel.border = element_blank(),
axis.line.y = element_line(),
panel.grid.major.x = element_blank(),
panel.grid.minor.x = element_blank(),
panel.grid.major.y = element_blank(), # 删除主要Y轴网格线
panel.grid.minor.y = element_blank(), # 删除次要Y轴网格线
plot.margin = unit(c(1, 1, 1, 1), "cm")
)
ggsave("G.png",plot = p,width = 12, height = 8,dpi=300,limitsize = FALSE)
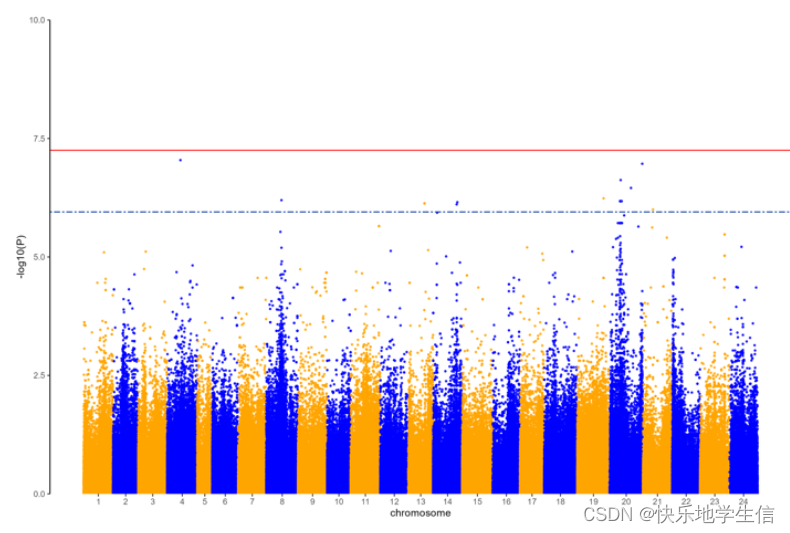
( 随便找的一个存库里面的图)
2.3CMplot画图
这里我没有太多尝试,但是网上用个CMplot画图的很多,当时我们想要矢量图,因为图片太大,想要降低分辨率其实作用不大,但是CMplot好像不能满足我们的条件,所以没有怎么尝试,这里给几个链接,下次有机会再试
GWAS分析中可视化:qqman和cmplot - 知乎 (zhihu.com)
总结一点,
使用R包最重要的是,
包的参数,多看各种教程的代码,找到合适的, 以调整自己喜欢的颜色和满足要求的格式为主。

















 1334
1334

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









