







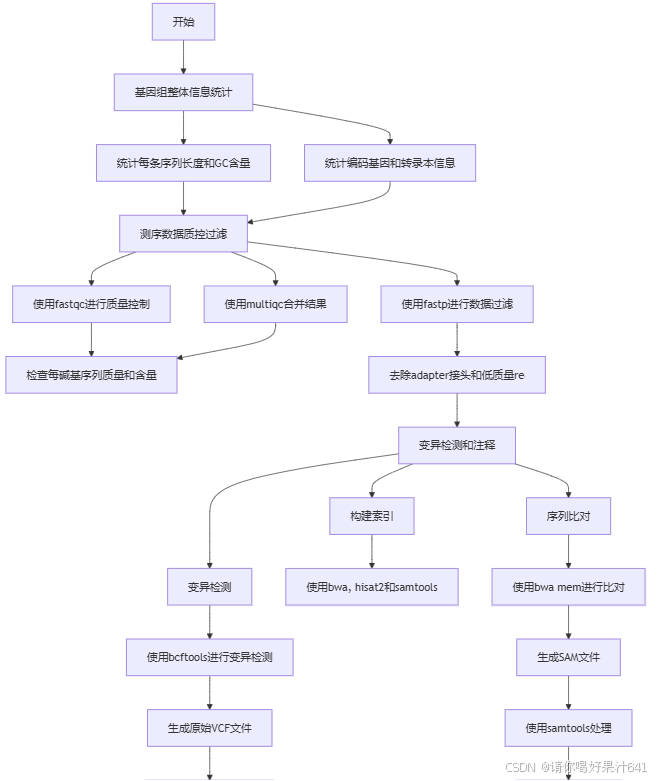
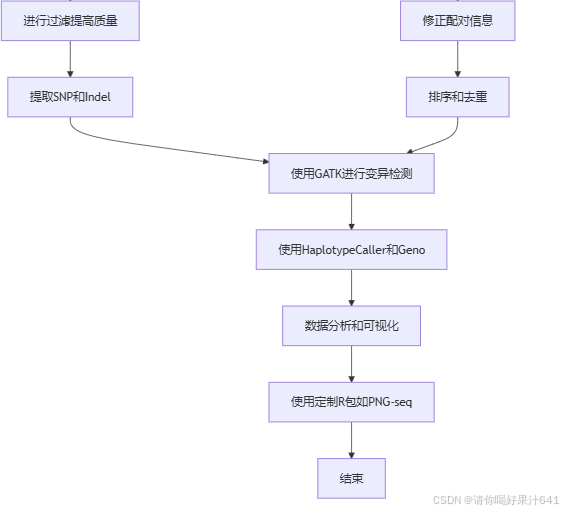
1.对了解参加基因组
1. 统计基因组整体信息
srun -A 2022099 -p Debug -n 1 -N 1 \
seqkit stats ~/yiyaoran/workspace/03.VariantCalling/01.ref/genome.fasta > genome.allstat
#srun: job 1272108 queued and waiting for resources
#srun: job 1272108 has been allocated resources
cat genome.allstat
file format type num_seqs sum_len min_len avg_len max_len
/public/home/2022099/yiyaoran/workspace/03.VariantCalling/01.ref/genome.fasta FASTA DNA 685 2,182,075,994 30,084 3,185,512.4 308,452,471
该文件 **`genome.allstat`** 是通过 **seqkit stats** 命令生成的,内容展示了参考基因组文件的一些统计信息。让我们逐项解释各列的含义:
### 文件内容解释:
- **file**:这是统计信息所属的文件。在此例中,`/public/home/2022099/yiyaoran/workspace/03.VariantCalling/01.ref/genome.fasta` 是被分析的参考基因组文件。
- **format**:文件的格式,这里为 **FASTA** 格式,它是常用的基因组或序列存储格式。
- **type**:序列的类型,这里是 **DNA** 类型,表示文件中包含的是 DNA 序列。
- **num_seqs**:序列的数量。此列表示在文件中有 685 条序列。
- **sum_len**:所有序列的总长度。这里表示文件中所有 685 条序列的总长度为 **2,182,075,994** 个碱基。
- **min_len**:序列的最小长度。表示在这些序列中,最短的序列长度为 **30,084** 个碱基。
- **avg_len**:序列的平均长度。表示文件中所有序列的平均长度为 **3,185,512.4** 个碱基。
- **max_len**:序列的最大长度。表示文件中最长的序列长度为 **308,452,471** 个碱基。
### 总结:
该文件展示了 **genome.fasta** 文件的基本统计信息。它包含 685 条 DNA 序列,\
总长度为约 21.8 亿个碱基,序列的长度从 **30,084** 到 **308,452,471** 个碱基不等\
···平均长度约为 **3,185,512.4** 个碱基。
目的:
使用 seqkit stats 对整个基因组的 FASTA 文件进行统计,得到基因组的基本信息,包括:
- 序列总数:染色体或其他序列的数量。
- 总碱基数:整个基因组的碱基总长度。
- 平均序列长度:各条染色体或序列的平均长度。
- 最短和最长序列:识别基因组中最短和最长的序列。
这些信息为研究人员提供了基因组的总体特征,便于了解基因组数据的规模和质量。
2. 统计每条序列长度
srun -A 2022099 -p Debug -n 1 -N 1 \
seqkit fx2tab -g -l -n -i ../genome.fasta > genome.seqstat
#srun: job 1272110 queued and waiting for resources
#srun: job 1272110 has been allocated resources
cat genome.seqstat |head -n 3
Seq_ID Length GC_Content
1 308452471 46.84
2 243675191 46.75
3 238017767 46.69
第一列:序列编号。这个编号表示参考基因组中的每条序列或染色体的编号。比如,1 是染色体 1,依此类推。
第二列:序列长度。这表示每条序列的总碱基数,例如,第 1 条序列(编号 1)长度为 308,452,471 个碱基,第 2 条序列(编号 2)的长度为 243,675,191 个碱基,依此类推。
第三列:GC 含量百分比。GC 含量是序列中碱基 G 和 C 的比例,表示为百分比。例如,第 1 条序列的 GC 含量为 46.84%,第 2 条序列的 GC 含量为 46.75%,依此类推。这表示在这些序列中,接近一半的碱基是 G 或 C。
目的:
使用 seqkit fx2tab 命令来逐条统计基因组中每个序列的详细信息:
- 序列名称:每条染色体或其他序列的名称。
- 序列长度:每条序列的长度。
这一步有助于研究人员了解基因组中每条染色体或 contig 的长度分布,便于评估不同染色体或组装片段的长度差异。如果存在非常短的序列,可能需要进一步检查数据质量。
3. 统计编码基因信息
# 使用制表符作为分隔符处理文件中的每一行
awk -F "\t" '
# 如果第3列等于"gene"并且最后一列包含"protein_coding"
$3 == "gene" && $NF ~ /protein_coding/ {
# 计数器num记录满足条件的基因数
num += 1;
# 计算每个基因的长度,并累加到total中
total += $5 - $4 + 1;
}
# 处理完所有行后执行以下语句
END {
# 输出基因的数量
print "number: " num;
# 输出所有基因的总长度
print "total_len: " total;
# 计算并输出平均基因长度
print "avg_len: " total / num;
}
' Oryza_sativa.IRGSP-1.0.53.gtf > gene.stat
目的:
使用 awk 脚本从 GTF 文件中提取编码基因的信息,统计如下内容:
- 编码基因的数量:统计 GTF 文件中被标记为“基因”(
gene)并且是蛋白质编码基因的条目数量。 - 总长度:计算这些蛋白质编码基因的总长度。
- 平均长度:计算每个基因的平均长度。
这一操作有助于了解基因组中的蛋白质编码基因的分布和结构特征,如基因数量、长度分布等,便于进一步分析基因组的功能性内容。
4. 统计编码转录本信息
# 使用制表符作为分隔符处理文件中的每一行
awk -F "\t" '
# 如果第3列等于"transcript"并且最后一列包含"protein_coding"
$3 == "transcript" && $NF ~ /protein_coding/ {
# 计数器num记录满足条件的转录本数
num += 1;
# 计算每个转录本的长度,并累加到total中
total += $5 - $4 + 1;
}
# 处理完所有行后执行以下语句
END {
# 输出转录本的数量
print "number: " num;
# 输出所有转录本的总长度
print "total_len: " total;
# 计算并输出平均转录本长度
print "avg_len: " total / num;
}
' Oryza_sativa.IRGSP-1.0.53.gtf > transcript.stat
目的:
使用 awk 脚本从 GTF 文件中提取编码转录本的信息,统计如下内容:
- 编码转录本的数量:统计 GTF 文件中被标记为“转录本”(
transcript)并且是蛋白质编码转录本的条目数量。 - 总长度:计算这些蛋白质编码转录本的总长度。
- 平均长度:计算每个转录本的平均长度。
转录本是基因表达的直接产物,了解转录本的信息有助于研究基因表达的复杂性,如同一个基因可能具有多个转录本,并且这些转录本长度和数量可以提供基因功能的多样性。
总体目的:
- 基因组的基本统计:通过基因组序列的统计信息,研究人员可以掌握基因组的规模、序列长度分布,以及可能存在的重复或缺失。
- 编码区域的特征统计:编码基因和转录本是基因组中功能性区域的代表。统计这些区域的数量和长度分布,有助于研究基因组的功能性内容,评估基因的密度、转录本的丰富性等,从而为后续的功能性分析打下基础。
- 数据质量控制:通过这些统计步骤,研究人员可以评估基因组和基因注释文件的完整性和准确性,检测是否存在异常情况,如过短的基因或转录本等。
这些统计步骤为研究人员提供了基因组的全貌,以及关于编码基因和转录本的详细信息,为后续的基因组学和转录组学分析提供了基础数据。
2.测序数据质控过滤
2.1 数据质控
fastqc \
--outdir ./qc \ # 输出目录为 ./qc,用于存放质量控制报告
--threads 4 \ # 使用 4 个线程进行并行处理,提高处理速度
../data/S1_1.fq.gz \ # 第一个样本的第一条读序列文件
../data/S1_2.fq.gz \ # 第一个样本的第二条读序列文件
../data/S2_1.fq.gz \ # 第二个样本的第一条读序列文件
../data/S2_2.fq.gz # 第二个样本的第二条读序列文件
#生成
./qc/B350H_L1_377X77.R1_fastqc.html ./qc/B350S_L1_384X84.R1_fastqc.html
./qc/B350H_L1_377X77.R1_fastqc.zip ./qc/B350S_L1_384X84.R1_fastqc.zip
./qc/B350H_L1_377X77.R2_fastqc.html ./qc/B350S_L1_384X84.R2_fastqc.html
./qc/B350H_L1_377X77.R2_fastqc.zip ./qc/B350S_L1_384X84.R2_fastqc.zip
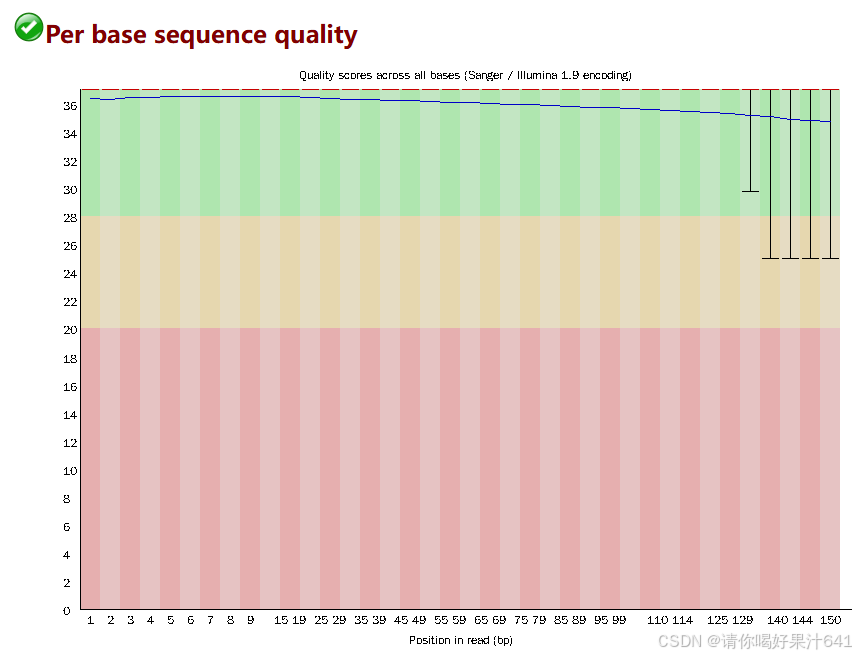
在报告中主要关注以下信息
Per base sequence quality(每碱基序列质量)
** Per base sequence content(每碱基序列含量)**
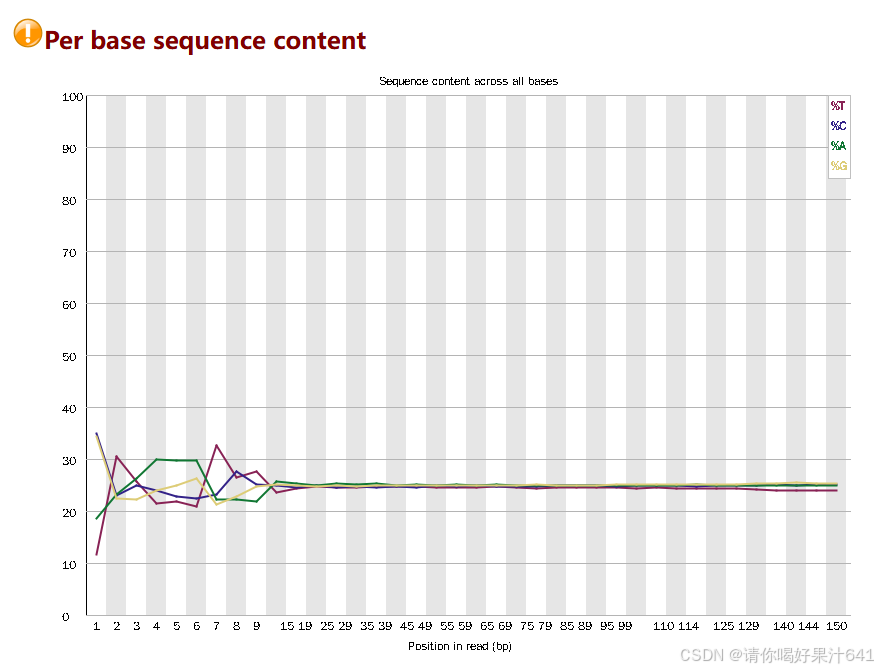
也可以使用mulitqc对qc结果进行合并
multiqc -d illumina_qc/ illumina_clean/:
#该命令的作用是从两个指定的文件夹 illumina_qc/ 和 illumina_clean/ 中读取质量控制结果,然后生成一个整合的报告。
过滤
-
Adapter接头:
- Adapter是用于连接目标DNA片段的接头序列,通常连接在目标序列的两端,确保测序仪能够捕捉到并读取DNA片段。
-
测序引物:
- 测序引物是用于启动DNA合成和测序反应的短序列片段,在Illumina等高通量测序平台上,测序引物与适配子区域结合,确保测序过程顺利进行。
-
Barcode:
- Barcode是一段独特的DNA序列,通常用于区分不同样本。在多样本的测序实验中,barcode允许将不同的样本混合在同一条通道中进行测序,最后再根据barcode序列进行分离和分析。
-
Index:
- Index与barcode类似,通常在多样本测序中用于区分不同的样本。Index也可以帮助标记不同的文库或实验。
适配子(adapter)的过程,主要解释了两种可能需要去除adapter的情况:
-
空载情况:
- 当适配子与适配子之间直接连接时,中间没有插入片段,导致测序读取时,Read 1 会测到 3’端的适配子,Read 2 则会测到 5’端适配子的反向互补区域。这种情况常发生在没有目标序列的文库或低浓度文库中。
-
插入片段过短情况:
- 如果插入的片段长度小于测序机器所能读取的循环次数(cycle 数),就会导致 Read 1 和 Read 2 的尾部都测到适配子序列。这种情况发生在文库片段较短时,适配子区域也会进入测序范围,影响后续分析。
去除包含过多“N”碱基的 reads 数据。
这里的 “N” 代表测序过程中无法确定的碱基,也就是说,测序仪无法准确读取这些位置的碱基信息。如果一个 read 中的 “N” 碱基过多,可能会影响后续的分析,因此需要将这些 read 过滤掉。
去除低质量reads的标准,具体内容如下:
-
以Q20作为判断标准:
- 使用Q20(测序质量值为20)作为判断标准。Q20代表测序错误率为1%,也就是说碱基的正确率为99%。
-
低于Q20碱基占总碱基的比例:
- 在测序的reads中,如果低于Q20的碱基占据一条reads的总碱基数的比例较高,这条reads将被认为是低质量的。
-
例如低于Q20的比例占30%:
- 如果某条reads中,低于Q20的碱基比例超过30%,这条reads就会被去除。这一阈值可以根据具体情况调整,用来过滤掉质量较差的reads。
duplication(重复序列)的原因通常有以下几点:
-
实验步骤中的PCR扩增:在建库过程中,PCR扩增可以增加某些序列的拷贝数,特别是在模板DNA浓度较低时,某些片段可能被过度扩增,导致重复读数的增加。
-
文库的低复杂度:如果样本文库的复杂度较低,测序仪器将重复读取相同的序列,从而导致duplication。低复杂度的文库可能是因为样本起始量较少或者文库制备过程中丢失了大量多样性。
-
测序深度过高:如果测序深度非常高,相比于文库的复杂度,重复序列的出现可能会增加,这意味着同一片段被测序多次。
去除duplication的原因:
-
去除冗余数据:duplication的存在可能会夸大某些区域的覆盖度,给下游数据分析带来偏差。去除重复序列可以让数据更准确反映样本中的真实情况。
-
提高数据质量:重复序列可能导致假阳性,影响变异检测和基因表达量的准确性。去除重复序列有助于提高测序数据的可靠性和分析结果的准确性。
-
优化计算资源:去除重复序列可以减少数据量,使得下游分析(如比对、变异检测等)更加高效。
数据处理原则,主要有两个要点:
-
不要求100%精确:在数据处理中,追求精确并不是绝对的目标。只要处理的原则不影响后续分析,数据的精度可以适当放宽。这意味着,少量误差是可以接受的。
-
根据最终结果重新过滤数据:在数据分析的过程中,可以根据分析结果的表现,决定是否需要重新调整和过滤数据。这种灵活性允许在不同分析阶段做出调整,以优化最终结果。
这两个原则帮助避免过于苛求数据处理中的每一步,灵活应对分析中的变化。
fastp \
--thread 4 \ # 使用4个线程并行处理数据
-i ../data/S1_1.fq.gz \ # 输入第一个样本的第一条读序列文件
-I ../data/S1_2.fq.gz \ # 输入第一个样本的第二条读序列文件
-o S1_1.fq.gz \ # 输出清洗后的第一条读序列文件
-O S1_2.fq.gz \ # 输出清洗后的第二条读序列文件
-j S1.fastp.json \ # 输出第一个样本的质量控制报告(JSON格式)
-h S1.fastp.html \ # 输出第一个样本的质量控制报告(HTML格式)
2>S1.fastp.log # 将过程中的日志输出到S1.fastp.log文件中
fastp \
--thread 4 \ # 使用4个线程并行处理数据
-i ../data/S2_1.fq.gz \ # 输入第二个样本的第一条读序列文件
-I ../data/S2_2.fq.gz \ # 输入第二个样本的第二条读序列文件
-o S2_1.fq.gz \ # 输出清洗后的第一条读序列文件
-O S2_2.fq.gz \ # 输出清洗后的第二条读序列文件
-j S2.fastp.json \ # 输出第二个样本的质量控制报告(JSON格式)
-h S2.fastp.html \ # 输出第二个样本的质量控制报告(HTML格式)
2>S2.fastp.log # 将过程中的日志输出到S2.fastp.log文件中
### 也可以这样
fastp \
-i illumina_1.fastq.gz \ # 输入的第一个fastq文件(illumina_1.fastq.gz)
-I illumina_2.fastq.gz \ # 输入的第二个fastq文件(illumina_2.fastq.gz)
-o clean.1.fq.gz \ # 输出过滤后的第一个fastq文件(clean.1.fq.gz)
-O clean.2.fq.gz \ # 输出过滤后的第二个fastq文件(clean.2.fq.gz)
-z 4 \ # 设置压缩输出的级别为 4
-q 20 \ # 设置质量过滤阈值为20,低于该阈值的碱基将被剪切
-u 40 \ # 允许最多40%的低质量碱基
-n 10 \ # 允许每条序列最多包含10个'N'碱基
-f 15 \ # 从每条序列的开头剪切掉15个碱基
-t 15 \ # 从每条序列的末尾剪切掉15个碱基
-F 15 \ # 剪切掉前端的15个碱基
-T 15 \ # 剪切掉末端的15个碱基
-D \ #duplication
-h fastp.html # 生成过滤过程的HTML格式报告文件(fastp.html)
3.变异检测和注释
3.1构建index
#bwa 构建 index, 用于DNA数据比对
bwa index genome.fasta
#hisat2 构建 index, 用于RNAseq数据比对
hisat2-build genome.fasta genome.fasta
#生成.fai 和 .dict文件, GATK软件依赖
samtools faidx genome.fasta
samtools dict -o genome.dict genome.fasta
#samtools 生成:
genome.dict
genome.fasta.fai
#bwa 生成
genome.fasta.amb
genome.fasta.ann
genome.fasta.bwt
genome.fasta.pac
genome.fasta.sa
#hisat2 生成
genome.fasta.1.ht2
genome.fasta.2.ht2
genome.fasta.3.ht2
genome.fasta.4.ht2
genome.fasta.5.ht2
genome.fasta.6.ht2
genome.fasta.7.ht2
genome.fasta.8.ht2
3.2比对
这里主要使用bwa进行比对(https://blog.csdn.net/2302_80012625/article/details/142903229?spm=1001.2014.3001.5502)
bwa mem -t 4 reference.fa sample_1.fq.gz sample_2.fq.gz > output.sam
# 使用 BWA 进行比对,并生成 SAM 文件
bwa mem -t 8 \ # 使用 8 个线程并行处理
-R '@RG\tID:H1\tSM:H1\tPL:illumina' \ # 添加 read group 信息:ID 为 H1,样本名为 H1,测序平台为 illumina
../01.ref/genome.fasta \ # 参考基因组文件(FASTA 格式) (已经通过 `bwa index` 构建)
/public/home/2022099/yiyaoran/workspace/02.ReadsFilter/H1_1.fq.gz \ # 第一条测序读取(FASTQ 格式,压缩)
/public/home/2022099/yiyaoran/workspace/02.ReadsFilter/H1_2.fq.gz \ # 第二条测序读取(FASTQ 格式,压缩)
-o H1.sam \ # 输出结果到 H1.sam 文件中
2>H1.bwa.log && # 将 BWA 比对过程中产生的日志输出到 H1.bwa.log 文件中
# 使用 samtools fixmate 处理配对比对信息
samtools fixmate -m H1.sam H1.fixmate.bam && # 将 H1.sam 修正后输出为 BAM 格式文件,并确保 mate 信息正确 (-m 选项)
# 对 BAM 文件进行排序
samtools sort -@ 2 -m 2G \ # 使用 2 个线程,分配每个线程 2G 内存进行排序
-o H1.fixmate.sort.bam \ # 将排序后的结果输出为 H1.fixmate.sort.bam
H1.fixmate.bam && # 输入修正后的 BAM 文件进行排序
# 使用 samtools markdup 标记重复比对
samtools markdup H1.fixmate.sort.bam H1.fixmate.sort.markdup.bam # 标记重复比对的片段,并将结果保存为 H1.fixmate.sort.markdup.bam
samtools index S1.fixmate.sort.markdup.bam \ # 使用 samtools 对最终的 BAM 文件建立索引,便于后续快速查找
&& samtools flagstat S1.fixmate.sort.markdup.bam \ # 统计 BAM 文件的比对状态信息,包括总 reads 数、比对成功 reads 数等
> S1.sort.markdup.bam.flagstat \ # 将 flagstat 的统计结果输出到 S1.sort.markdup.bam.flagstat 文件中
&& samtools coverage S1.fixmate.sort.markdup.bam \ # 统计 BAM 文件中的基因组覆盖率信息
> S1.sort.markdup.bam.coverage # 将覆盖率信息输出到 S1.sort.markdup.bam.coverage 文件中
3.3 bcftools 变异检测
ls ../step1_mapping/*.sort.markdup.bam > bam.list \ # 列出目录 ../step1_mapping 下所有以 .sort.markdup.bam 结尾的 BAM 文件,并将其文件列表保存到 bam.list 文件中
bcftools mpileup \ # 使用 bcftools mpileup 对 BAM 文件生成 pileup 信息
--bam-list bam.list \ # 指定 bam.list 文件,包含待处理的 BAM 文件路径
--annotate DP,AD,ADF,ADR \ # 注释生成的 VCF 文件,包含深度(DP),等位基因频率(AD),正链和负链的等位基因频率(ADF、ADR)
--threads 4 \ # 使用 4 个线程进行并行处理
--no-BAQ \ # 不使用 BAQ(基本比对质量)调整
--min-MQ 20 \ # 设置最低比对质量为 20,低于此值的比对将被忽略
--min-BQ 18 \ # 设置最低碱基质量为 18,低于此值的碱基将被忽略
--adjust-MQ 50 \ # 调整比对质量到 50
--output-type u \ # 输出未压缩的 VCF 格式
--fasta-ref ../01.ref/genome.fasta \ # 指定参考基因组文件
| \ # 管道操作,将输出传递给 bcftools call 命令
bcftools call \ # 使用 bcftools call 调用变异
--threads 4 \ # 使用 4 个线程并行处理
--multiallelic-caller \ # 使用多等位基因调用模式
--annotate GQ,GP \ # 注释生成的 VCF 文件,包含基因型质量(GQ)和基因型概率(GP)
--variants-only \ # 仅输出变异位点,忽略非变异位点
--output-type z \ # 输出压缩的 VCF 格式(gz 压缩)
--output all.raw.vcf.gz # 将结果输出到 all.raw.vcf.gz 文件中
####过滤
bcftools filter -i 'INFO/MQ>=40 && QUAL>20' \ # 使用过滤条件,过滤 MQ(比对质量)大于等于 40 且 QUAL(质量)大于 20 的变异位点
-O z \ # 输出为压缩的 VCF 格式
-o all.filter.vcf.gz \ # 将过滤后的结果保存到 all.filter.vcf.gz 文件中
all.raw.vcf.gz # 输入原始的未过滤的 VCF 文件 all.raw.vcf.gz
####分别输出snp、indel
bcftools view --types snps \ # 使用 bcftools view 只保留 SNP(单核苷酸多态性)变异
--output-type v \ # 输出为 VCF 格式
--output snp.filter.vcf.gz \ # 将 SNP 过滤后的结果保存到 snp.filter.vcf.gz 文件中
all.filter.vcf.gz # 输入已经过滤的 VCF 文件 all.filter.vcf.gz
bcftools view --types indels \ # 使用 bcftools view 只保留 Indel(插入/缺失)变异
--output-type v \ # 输出为 VCF 格式
--output indel.filter.vcf.gz \ # 将 Indel 过滤后的结果保存到 indel.filter.vcf.gz 文件中
all.filter.vcf.gz # 输入已经过滤的 VCF 文件 all.filter.vcf.gz
3.3GATK进行变异检测
GATK 变异检测流程总结
1. 准备工作
- 参考基因组:准备参考基因组文件,并使用工具(如
samtools faidx和samtools dict)为参考基因组创建索引。 - 样本数据:准备测序样本的比对结果(通常是 BAM 文件),这些文件需要经过质量控制、比对、排序和去重等处理。
2. HaplotypeCaller 进行单样本 GVCF 变异检测
- 目的:对于每个样本,使用 GATK 的
HaplotypeCaller工具进行变异检测,生成每个样本的 GVCF 文件。 - 过程:
- 如果基因组较小,可以直接针对整个基因组进行变异检测。
- 对于大基因组,建议分染色体或分区块进行变异检测,以提高计算效率并降低内存占用。
- 示例命令:
如果分染色体检测,可以在gatk HaplotypeCaller \ -R reference.fasta \ -I sample.bam \ -O sample.g.vcf.gz \ --ERC GVCF-L选项中指定染色体,例如-L chr1。
3. 多样本 GVCF 合并与 GenotypeGVCFs
- 目的:将多个样本的 GVCF 文件按染色体或区块合并,并进行群体的基因型调用。
- 过程:
- 使用
CombineGVCFs命令,将分染色体的 GVCF 文件合并成一个染色体级别的 GVCF 文件。 - 使用
GenotypeGVCFs进行群体基因型调用,生成群体的 VCF 文件。
- 使用
- 示例命令:
# 合并多个 GVCF 文件 gatk CombineGVCFs \ -R reference.fasta \ -V sample1.g.vcf.gz \ -V sample2.g.vcf.gz \ -O combined.g.vcf.gz # 基因型调用 gatk GenotypeGVCFs \ -R reference.fasta \ -V combined.g.vcf.gz \ -O combined.raw.vcf.gz
4. 合并染色体级别的 VCF 文件
- 目的:如果分染色体进行了变异检测,最后需要将各染色体的 VCF 文件合并成一个完整的全基因组 VCF 文件。
- 工具:可以使用 GATK 的
MergeVcfs命令或bcftools concat。 - 示例命令:
gatk MergeVcfs \ -I chr1.vcf.gz \ -I chr2.vcf.gz \ -O genome.vcf.gz
CombineGVCFs:用于 GVCF 文件 的合并,主要在 基因型调用前,将分染色体或多样本的 GVCF 文件合并,适用于后续使用 GenotypeGVCFs 命令进行基因型调用。将突变体与野生型的1号染色体合在一起
MergeVcfs:用于 VCF 文件 的合并,主要在 基因型调用后,将分染色体或分区块的 VCF 文件合并为一个完整的全基因组 VCF 文件,适用于最终的变异结果分析。讲各各染色体合在一起
5. 提取 SNP 和 Indel 并过滤
- 目的:从 VCF 文件中分别提取 SNP 和 Indel 变异,并对其进行质量过滤。
- 过程:
- 使用
bcftools或 GATK 的SelectVariants命令提取 SNP 和 Indel。 - 对变异进行过滤,通常依据质量评分(如 MQ、QUAL)。
- 使用
- 示例命令:
# 提取 SNP gatk SelectVariants \ -R reference.fasta \ -V genome.vcf.gz \ --select-type-to-include SNP \ -O snps.vcf.gz # 提取 Indel gatk SelectVariants \ -R reference.fasta \ -V genome.vcf.gz \ --select-type-to-include INDEL \ -O indels.vcf.gz # 过滤 gatk VariantFiltration \ -R reference.fasta \ -V snps.vcf.gz \ --filter-expression "QD < 2.0 || FS > 60.0" \ --filter-name "LowQual" \ -O filtered_snps.vcf.gz
总结
GATK 的变异检测流程包括以下几个主要步骤:
- HaplotypeCaller:对每个样本进行 GVCF 格式的变异检测,建议在大基因组时分染色体检测。
- CombineGVCFs 和 GenotypeGVCFs:将多个样本的 GVCF 文件合并后,进行群体基因型调用。
- 合并 VCF:如果分染色体进行变异检测,需要将分染色体的 VCF 文件合并成一个全基因组的 VCF 文件。
- 提取和过滤变异:分别提取 SNP 和 Indel 变异,并进行质量过滤,最终生成可靠的变异集。
前三步
前3步分染色体命令重复且多,写脚本完成
# 定义参考基因组路径
ref=~/yiyaoran/workspace/03.VariantCalling/01.ref/genome.fasta
# 创建临时目录,用于存放中间文件
mkdir ./tmp
#cat sample_bam.list
#H1 ~/yiyaoran/workspace/03.VariantCalling/02.mapping/H1.fixmate.sort.markdup.bam
#S1 ~/yiyaoran/workspace/03.VariantCalling/02.mapping/S1.fixmate.sort.markdup.bam
# 读取 sample_bam.list 文件中的样本名称和对应的 BAM 文件
cat sample_bam.list | while read sample bam
do
# 为每个样本创建一个目录
mkdir $sample
# 读取 chr.list 中的染色体列表,针对每个染色体执行 GATK HaplotypeCaller
#cat chr.list
#1
#2
#3
#4
#5
#6
#7
#8
#9
#10
cat chr.list | while read chr
do
# 输出 HaplotypeCaller 的命令,生成 g.vcf 文件
echo "gatk --java-options \"-Xmx10g -Djava.io.tmpdir=./tmp\" \
HaplotypeCaller -R $ref -I $bam -L $chr -ERC GVCF \
-O $sample/$sample.$chr.g.vcf.gz \
1>$sample/$sample.$chr.HC.log 2>&1 "
done > step1.$sample.HaplotypeCaller.sh # 将每个样本的 HaplotypeCaller 命令写入 shell 脚本
done
# 删除之前生成的 CombineGVCFs 和 GenotypeGVCFs 的 shell 脚本(如果存在)
#rm -f step2.CombineGVCFs.sh
#rm -f step3.GenotypeGVCFs_gvcf.sh
# 针对每个染色体,合并所有样本的 g.vcf 文件
cat chr.list | while read chr
do
# 从 sample_bam.list 中提取样本名称,生成每个染色体的 g.vcf 文件列表
cut -f 1 sample_bam.list | while read sample
do
echo $sample/$sample.$chr.g.vcf.gz # 输出每个样本的 g.vcf 文件路径
done > gvcf.$chr.list # 将每个样本的 g.vcf 文件路径写入 gvcf 文件
# 输出 GATK CombineGVCFs 命令,将所有样本的 g.vcf 文件合并
echo "gatk --java-options \"-Xmx10g -Djava.io.tmpdir=./tmp\" \
CombineGVCFs -R $ref -V gvcf.$chr.list -O $chr.g.vcf.gz \
1>$chr.CombineGVCFs.log 2>&1 " >> step2.CombineGVCFs.sh
# 输出 GATK GenotypeGVCFs 命令,将合并后的 g.vcf 文件进行基因分型
echo "gatk --java-options \"-Xmx10g -Djava.io.tmpdir=./tmp\" \
GenotypeGVCFs -R $ref -V $chr.g.vcf.gz -O $chr.raw.vcf.gz \
1>$chr.GenotypeGVCFs.log 2>&1 " >> step3.GenotypeGVCFs_gvcf.sh
done
生成脚本如下
##step1
gatk --java-options "-Xmx10g -Djava.io.tmpdir=./tmp" \
HaplotypeCaller \
-R /public/home/2022099/yiyaoran/workspace/03.VariantCalling/01.ref/genome.fasta \ # 引用参考基因组
-I ~/yiyaoran/workspace/03.VariantCalling/02.mapping/H1.fixmate.sort.markdup.bam \ # 输入 BAM 文件
-L 2 \ # 指定染色体 2
-ERC GVCF \ # 输出 GVCF 格式
-O H1/H1.2.g.vcf.gz \ # 输出文件路径及文件名
1>H1/H1.2.HC.log 2>&1 # 将标准输出和错误日志写入 log 文件
。。。。。。。。。不再重复
##step2
#cat gvcf.1.list
#H1/H1.1.g.vcf.gz
#S1/S1.1.g.vcf.gz
gatk --java-options "-Xmx10g -Djava.io.tmpdir=./tmp" \
CombineGVCFs \
-R /public/home/2022099/yiyaoran/workspace/03.VariantCalling/01.ref/genome.fasta \ # 参考基因组路径
-V gvcf.1.list \ # 输入 GVCF 文件列表,包含染色体 1 的所有样本
-O 1.g.vcf.gz \ # 输出合并后的 GVCF 文件
1>1.CombineGVCFs.log 2>&1 # 将日志文件保存到 1.CombineGVCFs.log
。。。。。。。。。不再重复
##step3
gatk --java-options "-Xmx10g -Djava.io.tmpdir=./tmp" \
GenotypeGVCFs \ # 使用 GenotypeGVCFs 进行基因型调用
-R /public/home/2022099/yiyaoran/workspace/03.VariantCalling/01.ref/genome.fasta \ # 参考基因组文件路径
-V 1.g.vcf.gz \ # 输入 GVCF 文件,该文件是前一步合并的染色体 1 的 GVCF 文件
-O 1.raw.vcf.gz \ # 输出文件名,生成基因型调用后的 VCF 文件
1>1.GenotypeGVCFs.log 2>&1 # 将标准输出和标准错误日志保存到日志文件中
。。。。。。。。。不再重复
第4步合并染色体级别的 VCF 文件
##step4
#cat raw_vcf.list
#1.raw.vcf.gz
#2.raw.vcf.gz
#3.raw.vcf.gz
#4.raw.vcf.gz
#5.raw.vcf.gz
#6.raw.vcf.gz
#7.raw.vcf.gz
#8.raw.vcf.gz
#9.raw.vcf.gz
#10.raw.vcf.gz
gatk --java-options "-Xmx10g -Djava.io.tmpdir=./tmp" \
MergeVcfs \ # 使用 MergeVcfs 命令合并多个 VCF 文件
-I raw_vcf.list \ # 输入 VCF 文件列表,其中列出了要合并的 VCF 文件(通常是不同染色体的 VCF 文件)
-O all.merge_raw.vcf # 输出合并后的 VCF 文件,包含整个基因组的变异信息
第五步
gatk做硬筛参考(https://blog.csdn.net/2302_80012625/article/details/142879484?spm=1001.2014.3001.5501)
1. 提取 SNP 变异
gatk --java-options "-Xmx4g -Djava.io.tmpdir=./tmp" \
SelectVariants \ # 使用 SelectVariants 工具,从 VCF 文件中提取特定类型的变异
-R ../01.ref/genome.fasta \ # 指定参考基因组文件
-V all.merge_raw.vcf \ # 输入合并后的全基因组 VCF 文件
--select-type SNP \ # 选择变异类型为 SNP(单核苷酸多态性)
-O all.raw.snp.vcf # 输出提取后的 SNP 变异文件
2. 过滤 SNP 变异
gatk --java-options "-Xmx4g -Djava.io.tmpdir=./tmp" \
VariantFiltration \ # 使用 VariantFiltration 工具对 SNP 变异进行过滤
-R ../01.ref/genome.fasta \ # 指定参考基因组文件
-V all.raw.snp.vcf \ # 输入提取的 SNP 变异文件
--filter-expression "QD < 2.0 || MQ < 40.0 || FS > 60.0 || SOR > 3.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0" \ # 定义过滤条件
--filter-name 'SNP_filter' \ # 为这些过滤条件命名
-O all.filter.snp.vcf # 输出过滤后的 SNP 文件
3. 排除已过滤的变异,获取通过过滤的 SNP
gatk --java-options "-Xmx4g -Djava.io.tmpdir=./tmp" \
SelectVariants \ # 使用 SelectVariants 工具排除已过滤的变异
-R ../01.ref/genome.fasta \ # 指定参考基因组文件
-V all.filter.snp.vcf \ # 输入过滤后的 SNP 文件
--exclude-filtered \ # 排除已被标记为过滤掉的 SNP
-O all.filtered.snp.vcf # 输出通过过滤的 SNP 变异文件
之后就是可视化用的PNG-seq这个R包https://github.com/smilejeen/PNGseqR


















 1810
1810

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









