







单核测序解析水稻雌蕊的发育轨迹
摘要
被子植物的生命周期包含孢子体和配子体世代的交替,这一过程发生在如雌蕊等植物器官中。水稻雌蕊包含胚珠并接收花粉以实现成功受精,从而产生种子。然而,目前对水稻雌蕊的细胞表达特征知之甚少。在本研究中,我们通过基于液滴的单核RNA测序(snRNA-seq)展示了水稻雌蕊在受精前的细胞普查。通过原位杂交验证的 ab initio 标记物识别,有助于细胞类型的注释,揭示了胚珠和心皮来源细胞之间的异质性。
1N(配子体)与2N(孢子体)细胞核的比较解析了胚珠中生殖细胞的发育路径,发现了在孢子体-配子体转换前典型的多能性重置现象。同时,对心皮来源细胞的轨迹分析揭示了表皮特化和花柱功能等先前未被关注的特征。这些发现提供了关于水稻雌蕊在开花前的细胞分化和发育的系统性视角,并为理解植物的雌性生殖发育奠定了基础。
引言
水稻(Oryza sativa)是最重要的谷物作物之一,特别是在亚洲地区,因为它的种子被当作主食消费。在一粒稻谷中,种子是一种颖果,这是一种单粒果实,其中果皮与种皮融合。这样的颖果源自雌性生殖器官——雌蕊,它由两个合生的花被片组成,内含一个胚珠,胚珠中承载着胚囊。成熟的水稻雌蕊结构包含一个瓶状的子房在基部,顶部有两个羽毛状的柱头,以及一个连接的分枝风格。子房内含有一个被子房壁包围的胚珠。在胚珠中,配子体细胞(1N;单倍体细胞)构成了嵌入在胚珠中心细胞部分的胚囊,这部分被称为珠心细胞,它们进一步被外保护层珠被覆盖。在受精和随后的种子发育过程中,珠心细胞被消耗,而珠被衍生的种皮和子房壁衍生的果皮被挤压在一起,形成了成熟种子的融合覆盖层。
之前的研究表明,在花分生组织中,雌蕊的发育特征已经被描绘出来。提出的“ABCDE”模型主要基于两种模式真双子叶植物的研究,拟南芥(Arabidopsis thaliana)和金鱼草(Antirrhinum majus)。C类基因和E类基因共同决定了花分生组织中最内层(第四轮)的花被片身份,而D类基因则在花被片内指定胚珠。在水稻早期雌蕊发育过程中,MADS-box转录因子OsMADS13作为“D类基因”的同源物,决定了胚珠的身份并抑制了花被片的身份。同时,在围绕胚珠原基的两个原基中,YABBY同源物DROOPING LEAF(DL)和两个C类基因,OsMADS3和OsMADS58,决定了花被片的身份。尽管水稻和拟南芥在雌蕊发育的分子遗传基础上有一些相似之处,但它们在雌蕊的发育轨迹和最终结构上表现出差异。在拟南芥中,多个胚珠原基从已建立的花被片的胎盘中出现,而在水稻雌蕊中,一个胚珠原基和两个周围的花被片原基几乎同时出现,分别发育成含有配子体的胚珠和雌蕊的其他部分。
(1. ABCDE模型描述了五类基因(A、B、C、D和E)通过特定表达模式调控花器官的分化,其中A类决定花萼,A+B类决定花瓣,B+C类决定雄蕊,C类决定心皮,D类决定胚珠,E类基因协同作用于所有花器官的发育。
2. 该模型揭示了基因表达区域的互相排斥和协调作用,为理解被子植物花器官从未分化组织到特化结构的形成提供了理论框架。
3. 在水稻中,C类基因(OsMADS3/58)与E类基因共同决定心皮的发育,D类基因(OsMADS13)特化胚珠,说明ABCDE模型在单子叶植物中同样适用。)
由于水稻雌蕊包含不同类型的细胞,使用新兴的高通量工具在单细胞水平上分析不同细胞类型对于阐明参与关键生殖过程的雌蕊的发育轨迹是必要的,这些过程从花粉识别到受精和种子形成。单细胞RNA测序(scRNA-seq)是一个选项,但它需要生成没有细胞壁的原生质体。因此,scRNA-seq的应用目前仅限于那些容易生成原生质体的某些营养器官,如根尖、12-23的茎尖分生组织、24的愈伤组织、25和年轻叶片。26-28由于许多植物组织,包括雌蕊,对原生质体化有抵抗力,一个替代策略是单核RNA测序(snRNA-seq),它快照了细胞状态,并避免了原生质体化过程中异常转录的可能性。此外,snRNA-seq捕获了细胞核中转录调控的确切信息,受到细胞质中其他细胞过程的干扰最小。迄今为止,成功应用snRNA-seq的样本与scRNA-seq的样本在很大程度上重叠,如拟南芥的根和幼苗29-31以及玉米(Zea mays)的幼苗和叶片剥离。32
在这项研究中,我们应用了一种改良的基于液滴的单核RNA测序(snRNA-seq)技术,以获得对原生质体化有抵抗力的水稻雌蕊的细胞普查,这种抵抗力部分是因为与其他器官相比,水稻雌蕊中富含果胶——一种异质性分支的多糖混合物。33我们进行了从头标记识别和细胞类型注释,以识别单细胞水平上雌蕊的整体细胞格局。我们的发现提供了迄今为止未知的水稻雌蕊细胞异质性和发育轨迹的视角,并为植物雌性生殖发育提供了洞见。
结果:
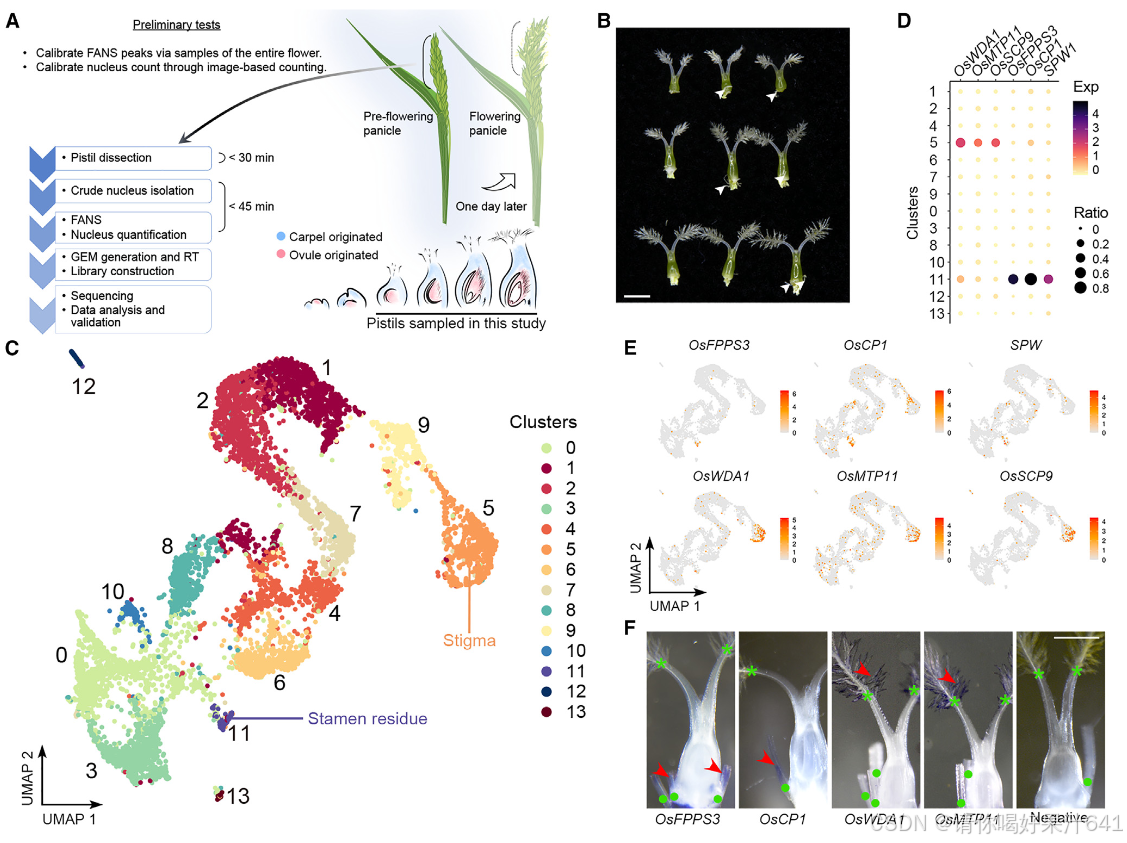
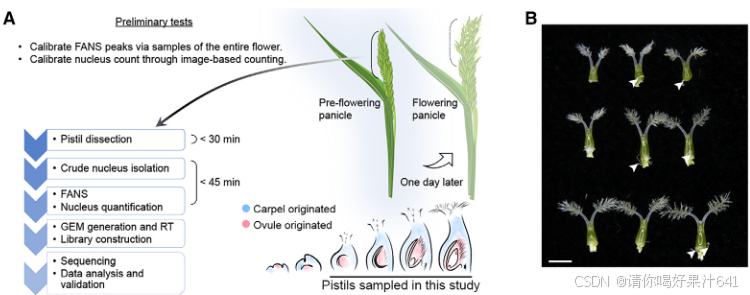
单核RNA测序(snRNA-seq)用于纯化水稻雌蕊中的细胞核
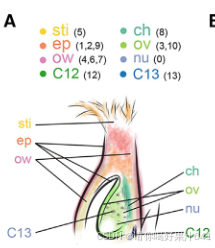
为了在单细胞水平上研究水稻雌蕊的细胞特征,我们首先为在开花前一天收集的未受精雌蕊的snRNA-seq开发了一个定制的工作流程(图1A)。从未开放的穗状花序中解剖出连续发育的雌蕊,并用液氮快速冷冻以最小化基因表达的变化,紧接着在短时间内通过荧光激活的细胞核排序(FANS)分离粗细胞核并进一步纯化。
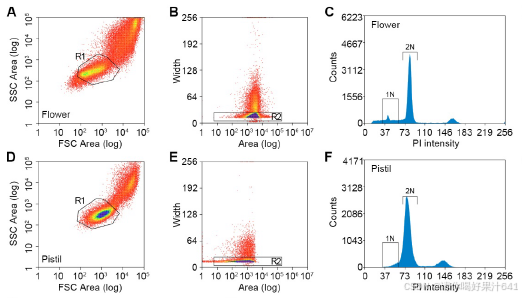
在这个工作流程中,我们解决了在构建snRNA-seq文库时遇到的两个实验挑战,包括对高质量细胞核的要求、精确的细胞核计数以及保持细胞核RNA的完整性。首先,我们优化了工作流程中使用的RNase抑制剂的浓度,以最小化RNA降解。其次,我们还进行了初步测试,以检查细胞核的质量并建立计数方法,以简化细胞核计数步骤。为此,我们通过细胞分选器分析了开放花朵中的细胞核,记录了被碘化丙啶(PI)染色荧光标记的2N(二倍体;孢子体细胞)和1N(单倍体;主要由花粉贡献)细胞核的峰值位置(图S1A-S1C)。从FANS分离出的细胞核是纯净且完整的(图S1G和S1H)。此外,从分选器和基于图像的方法得到的2N和1N细胞核计数结果的相关性(图S1H-S1J)允许我们根据分选器记录估算细胞核数量,以缩短工作流程所需的时间。
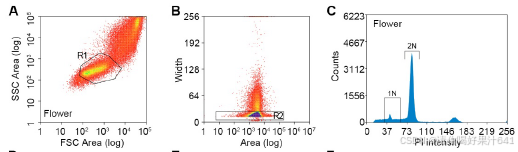
【【(A-F) 从花朵(A-C)和雌蕊(D-F)中分离的细胞核的分选策略。通过群体确定(A和D中的R1门)和单体型识别(B和E中的R2门)选择单个细胞核。从R1和R2门筛选出的细胞核根据碘化丙啶(PI)染色的荧光绘制直方图(C和F)。直方图中标示的假定1N或2N细胞核被分选到不同的试管中。】】
然后我们将已建立的工作流程应用于对开花前一天从穗状花序中收集的未受精雌蕊进行单核RNA测序(snRNA-seq)。在荧光激活的细胞核排序(FANS)过程中,我们将雌蕊2N细胞核的峰值与从花朵样本中获得的标准2N峰值对齐,而雌蕊1N细胞核的信号被2N峰值的尾部所掩盖(图S1A-S1F)。因此,我们根据假设的位置(图S1F)对2N细胞核和潜在的1N细胞核进行了排序,并分别进行了单核RNA测序的文库制备(图1A)。
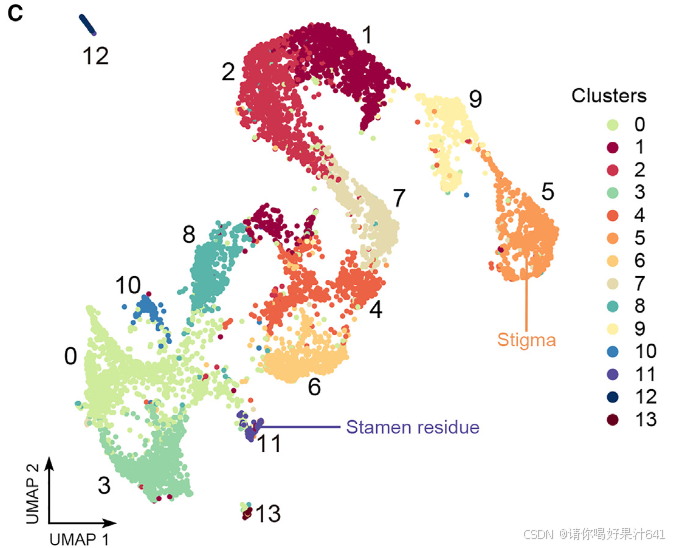
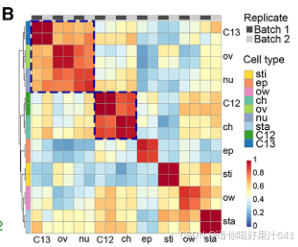
构成主要核群的2N细胞核随后被用于基于液滴的单核RNA测序(snRNA-seq),该测序通过10x Genomics平台处理。根据DoubletFinder的预测,我们去除了潜在的双细胞(doublets)。34我们进一步去除了低质量的细胞核,如果它们的超过5%或10%的读数分别映射到叶绿体或线粒体,以及检测到的基因数量超出300-3,000的范围。在应用这些质量过滤器后,我们总共获得了6,004个细胞核(第一批)和24,656个可检测基因(每个细胞759个)。我们还对另一个独立的生物学重复样本进行了snRNA测序,并获得了4,360个细胞核(第二批)和22,738个可检测基因(每个细胞455个),通过相同的协议进行过滤。第一批和第二批的数据表现出强烈的伪批量基因表达相关性(图S2A)。考虑到不同的数据质量(图S2B),我们随后根据独特分子标识符(UMI)分布23,35从第二批中选择了2,169个高质量细胞核,并将其与第一批数据结合进行进一步分析(总共8,173个细胞核;每个细胞700个)。通过Seurat 4.0(Hao等人36)对合并数据集进行的无监督分析揭示了14个不同的簇(0-13)(图1C),第一批和第二批的数据表现出类似的聚类趋势(图S2C)。为了验证数据质量,我们还使用RNA模板5’端的切换机制(SMART)技术,独立分析了通过相同程序为snRNA-seq分离的大量细胞核的转录组,并发现合并的snRNA-seq数据与大量细胞核RNA-seq数据之间存在高度相关性(图S2C和S2D),这表明我们的snRNA-seq数据中很好地保留了生物学信息。
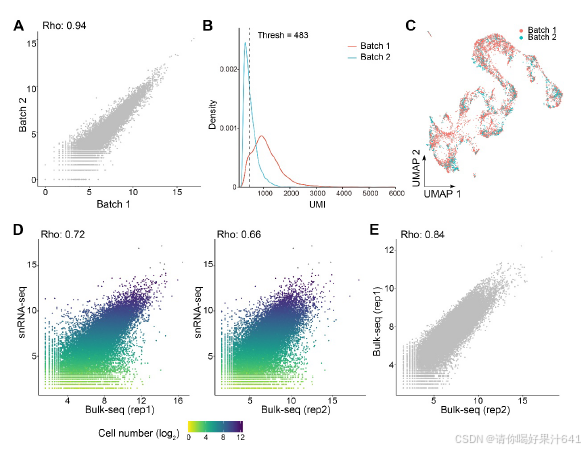
【【图S2. 比较两批snRNA-seq数据以及snRNA-seq数据与核批量seq数据之间的相关性。与图1相关。
(A) 第1批和第2批细胞的伪批量基因表达之间的相关性。伪批量基因表达是通过汇总所有细胞中一个基因的表达获得的。基因表达以log2尺度显示,相关性是按Spearman秩相关系数(Rho)计算的。
(B) 密度图显示第1批和第2批细胞每个细胞的总唯一分子标识符(UMIs)分布。
© 通过Seurat合并的第1批和第2批细胞的UMAP。在将高质量第2批细胞投影到结合两批数据的集成UMAP结构后,可视化了第1批和第2批细胞的UMAP结构。
(D) 集成snRNA-seq和批量核RNA-seq样本之间的基因表达相关性。点代表基因,颜色代表基因表达的细胞数量的log2数。集成snRNA-seq样本的伪批量基因表达是通过汇总所有细胞中一个基因的表达获得的。基因表达以log2尺度显示,相关性是按Spearman秩相关系数(Rho)计算的。
(E) 批量核RNA-seq重复之间的基因表达相关性。基因表达以log2尺度显示,相关性是按Spearman秩相关系数(Rho)计算的。
】】
2. 已知标记物对snRNA-seq的功能验证
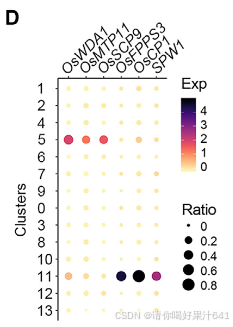
然后我们通过Seurat 4.0(表S1;详见STAR方法)为每个簇定义了潜在的标记基因。第11簇的标记基因包括B类基因SUPERWOMAN1(SPW1),这是一个雄蕊(第三轮)的身份基因,8表明第11簇可能代表在样本准备过程中难以消除的雄蕊残留物(图1B和1C)。整个样本的原位杂交证实了第11簇的其他两个潜在标记基因(OsFPPS3和OsCP1)确实在雄蕊残留物中表达(图1D-1F)。
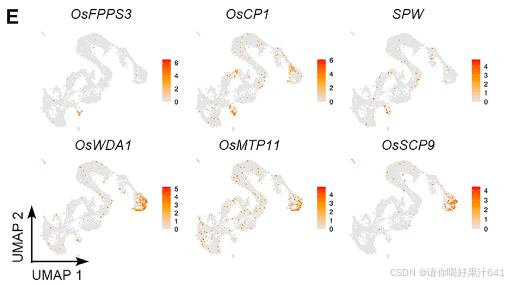
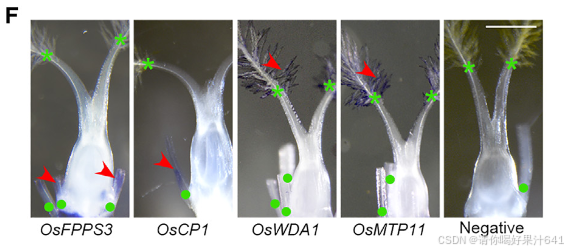
为了给后续的聚类识别提供一个大致的参考,我们将完整的雌蕊分为上部和下部(图S3A),这两部分都进行了RNA测序分析。这两个部分展现了可区分的表达谱(图S3B),表明空间上分离的部分存在遗传差异。我们随后比较了雌蕊上部或下部与每个簇的潜在标记基因之间的差异表达基因(DEGs)。在上部富集的基因与第5和第7簇的标记基因高度重叠,而下部富集的基因则显示出互补的模式(图S3C)。我们还比较了之前报道的潜在柱头特异性基因37与我们的标记基因列表,发现这些基因专门富集在第5簇(图S3C)。这些观察结果表明,第5簇包含柱头细胞,这与通过整体原位杂交验证的第5簇中两个潜在标记基因(OsWDA1和OsMTP11)在羽毛状柱头的表达一致(图1D-1F)。
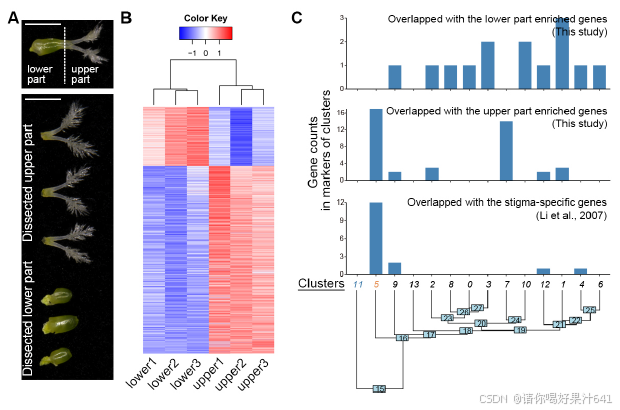
【【图S3. 使用批量RNA-seq比较簇标记。
与图1和图2相关。
(A) 批量RNA-seq的样本准备。雌蕊被分离成上部和下部进行RNA-seq分析。
(B) 热图比较雌蕊上部(三个重复样本)和下部(三个重复样本)差异表达基因(DEG)的表达。颜色代表按行标准化的log2读数计数。
© 与通过批量RNA-seq验证的组织特异性基因或报道的柱头特异性基因重叠的簇特异性标记基因的数量。基于Seurat的“BuildClusterTree”函数在PCA基因表达空间构建的距离矩阵估计了系统发育树。
】】
此外,我们还将之前报道的柱头特异性基因与我们的标记基因列表进行了比较,发现这些基因仅富集于簇5(图 S3C)。这一发现表明簇5包含柱头细胞,这一点与簇5的两个潜在标记基因(OsWDA1 和 OsMTP11)在羽毛状柱头中通过全组织原位杂交验证的表达结果一致(图1D–1F)。
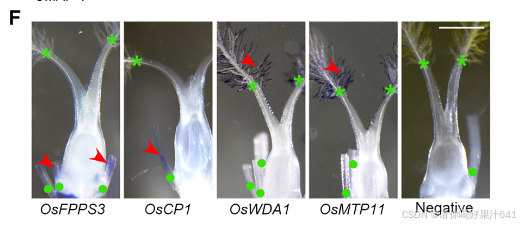
我们进一步通过Seurat 4.0构建了所有簇的系统发育树,并发现第11簇是最远缘相关的群体,其次是第5簇(图S3C)。这是意料之中的,因为雄蕊残留物不属于雌蕊,而柱头是雌蕊最上部的组织。因此,我们分别将第11簇和第5簇注释为雄蕊残留物和柱头(图1C)。其他簇应该代表构成雌蕊下部的不同细胞类型,包括子房和基部风格(图S3A)。这些结果支持我们的snRNA-seq数据聚类在生物学上是有意义的,可以用于进一步的分析。
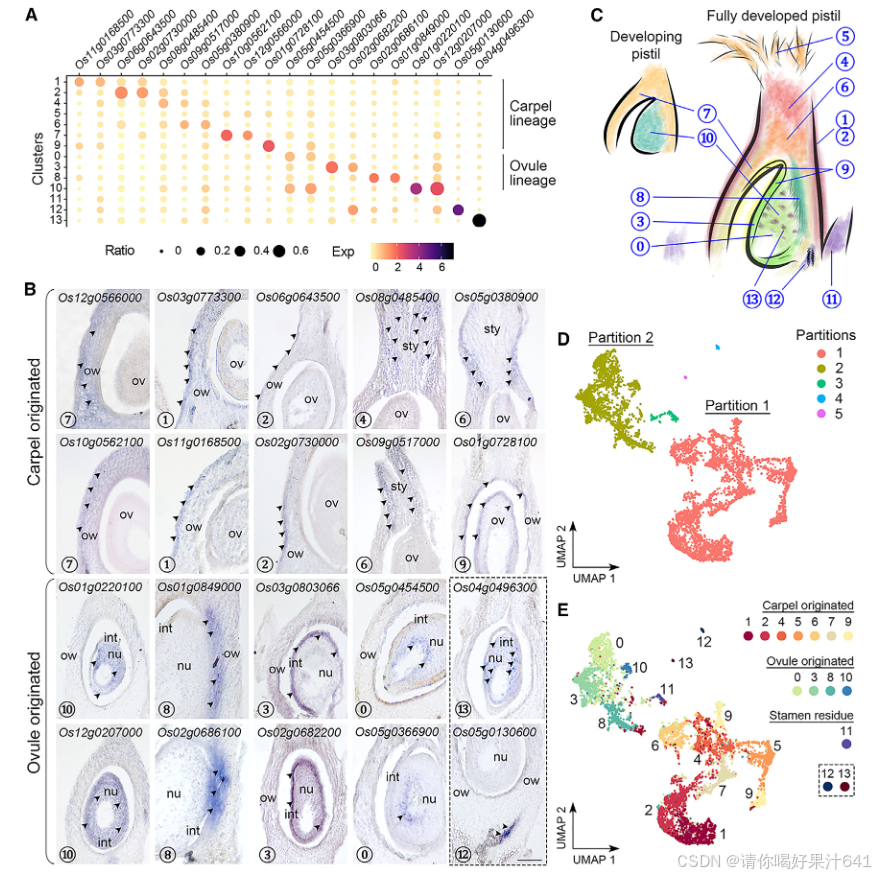
图2. 从头标记识别和验证
(A) 除了簇5和11之外的簇的标记基因表达。点的直径表示表达特定基因的簇细胞的比例(比率),而颜色表示该簇中细胞的平均表达(Exp)。图S4显示了每个细胞中标记基因表达谱的UMAP可视化。(B)通过原位杂交验证标记基因。蓝紫色-棕色染色,显示目标mRNA的存在,由箭头指示。每个面板底部的圆圈编号表示标记基因的Seurat聚类归属。比例尺,100毫米。int,珠被;nu,胚珠;ov,子房;ow,子房壁;sty,花柱。(C)Seurat聚类空间分布的卡通总结。(D)用Monocle3处理的细胞的UMAP可视化,点表示单个细胞(n = 8,173个细胞),颜色表示细胞分区。降维和分区识别由Monocle3执行。(E)Monocle3识别的主要分区反映了心皮和胚珠起源的细胞。细胞的颜色代表了用Seurat注释的聚类归属。分区1和2的簇(除了簇11-13)分别被指定为心皮和胚珠起源的细胞。
3. 对簇标记物的原始细胞类型注释
由于到目前为止还没有可用于水稻雌蕊中其他簇的经过验证表达的特定标记,我们试图通过原位杂交验证一些相应候选标记的时空定位来区分这些簇(图2A和S5;表S1)。
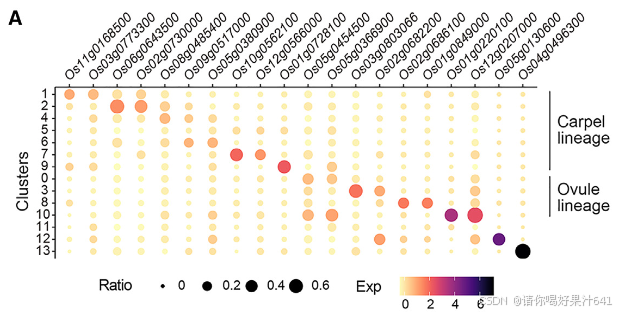
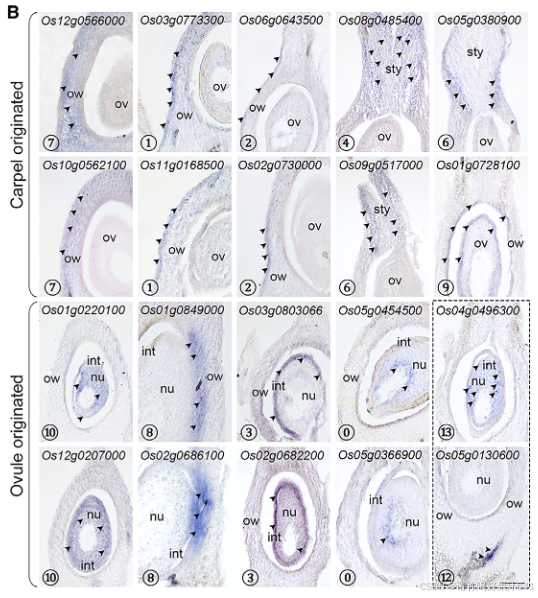
一组不同簇的标记基因主要在包围胚珠的子房和基部风格中表达(图2A、2B和S4)
- 簇7 的标记基因(Os10g0562100/OsMYBS2 和 Os12g0566000/OsBOR1)在子房壁中表达。
- 簇1和簇2 的标记基因(Os11g0168500/OsERF118、Os03g0773300、Os02g0730000/OsALDH2a 和 Os06g0643500/OsHyPRP18)在子房壁外表面中表达。
- 簇4 的标记基因(Os08g0485400)在花柱中检测到。
- 簇6 的标记基因(Os05g0380900/OsCML15 和 Os09g0517000)在子房顶端的基部花柱两侧检测到。
- 簇9 的标记基因(Os01g0728100/OsGELP24)在子房壁内表面和胚珠的外珠被中均有检测到。
这些结果表明,各簇标记基因在水稻雌蕊中具有特定的空间表达,为其细胞类型的注释提供了依据。
另一组标记基因的表达特异性定位于胚珠的不同部分(图2A、2B和S4):
- 簇10 的标记基因(Os12g0207000/OsMADS13 和 Os01g0220100/OsCEL9A)在整个胚珠中表达。
- 簇8 的标记基因(Os01g0849000/OsLTPd5 和 Os02g0686100)在胚珠的合点末端及其相连的子房壁中表达。
- 簇3 的标记基因(Os03g0803066/ncRNA 和 Os02g0682200/MFO1)在珠被中富集表达。
- 簇0 的标记基因(Os05g0454500 和 Os05g0366900)特异性表达在珠心的最内层。
- 簇13 的标记基因(Os04g0496300)仅在胚珠中检测到分散的点状表达,可能表明胚珠细胞处于某种特定状态。
- 簇12 的标记基因(Os05g0130600)在胚珠下方的维管组织中表达(图2B)。
综合分析
综合来看,除了簇5和簇11以外,我们对其他簇进行了 ab initio 注释,这些簇的表达覆盖了水稻雌蕊的不同区域(图2C)。这些发现为进一步研究水稻雌蕊中各细胞类型的功能特性提供了重要基础。
4.两大主要分区:心皮和胚珠来源的细胞
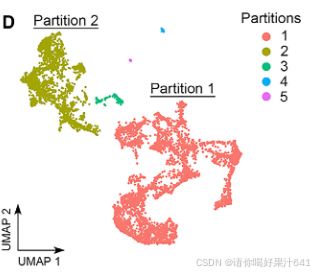
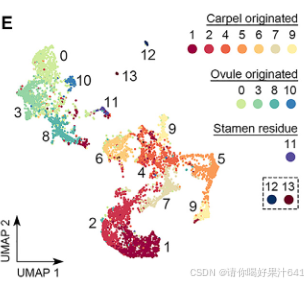
值得注意的是,使用 Monocle3(Cao 等人,2020)进行的分区分析将这些簇分为五个互不重叠的分区(图2D):
- 最大的分区 #1 包含簇1、2、4、5(柱头)、6、7和9。
- 较小的分区 #2 包含簇0、3、8和10(图2E)。
- 其他三个分区 #3、#4 和 #5 分别由簇11(雄蕊残余)、簇12和簇13组成,与前两个主要分区相比,这些分区仅包含极少量细胞核(图2D和2E)。
那些已知的花器官身份基因在花发育的早期阶段表达,而在本研究中检测的雌蕊中,它们中的大多数并没有被识别为特定簇的标记基因。然而,这些基因仍然可以被检测到,并且优先在两个主要分区(#1和#2)中表达。例如,
-
C类基因(OsMADS3/58)及其上游调控基因 OsMADS1 和心皮特异性基因 DL 主要富集于分区#1,尤其在簇7细胞中(图S5A–S5D)。
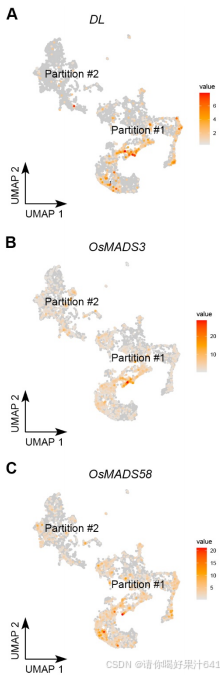
-
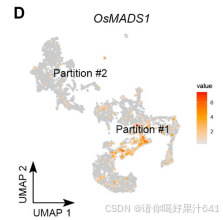
D类基因(OsMADS13)存在于分区#2,并被 Seurat 计算为簇10的标记基因(图S5E)。
-
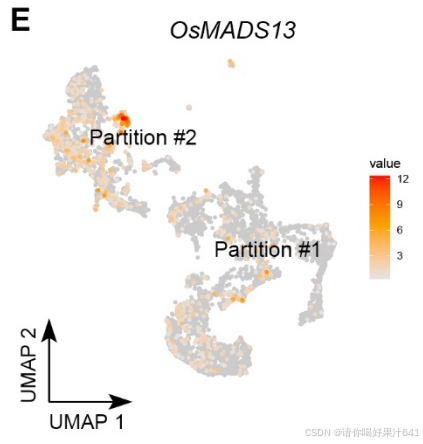
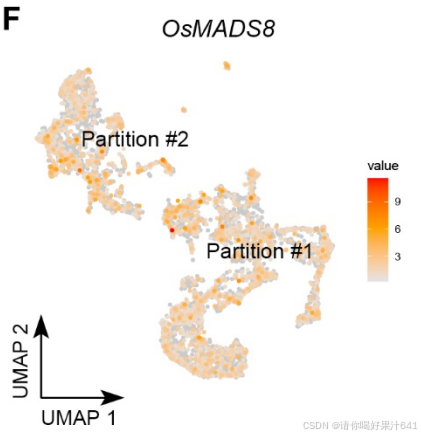
-
SEP3家族的E类基因(OsMADS8)控制C类和D类基因的表达,均匀分布于分区#1和#2(图S5F)。
结论
结合原位杂交结果,这些数据表明:
- 分区#1(簇1、2、4、5、6、7和9)代表心皮来源的细胞。
- 分区#2(簇0、3、8和10)代表胚珠来源的细胞。
此外,花器官特异性基因在簇7和簇10中的高表达表明,这两个簇包含发育中雌蕊中分化程度较低的细胞(图2C)。这进一步揭示了雌蕊发育过程中心皮和胚珠细胞的分区及功能特性。
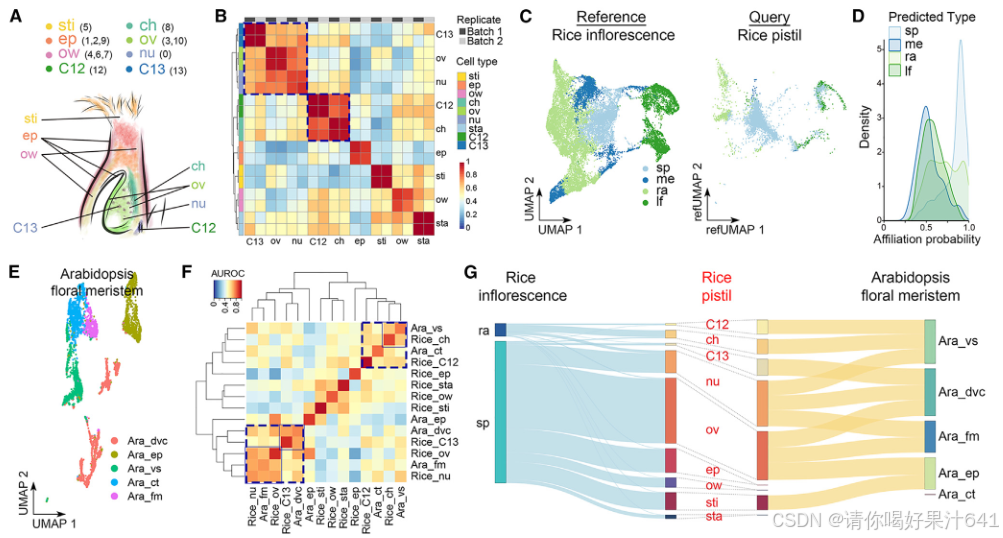
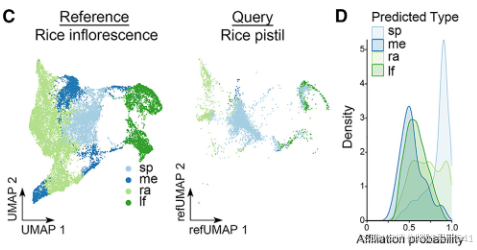
图3. 雌蕊、花序和花分生组织的细胞普查比较
(A) 基于水稻雌蕊中验证的空间表达信息的细胞类型注释。用数字表示的Seurat聚类被整合到由不同颜色代表的不同细胞类型中。ch,合点;ep,表皮;nu,胚珠;ov,子房;ow,子房壁;sti,柱头;C12,簇12细胞;C13,簇13细胞。
B)MetaNeighbor分析显示两批样本之间细胞类型的可复制性。
(C)将水稻雌蕊细胞(查询)投影到Seurat展示的水稻花序细胞的UMAP结构上。颜色代表不同的细胞类型。每个雌蕊细胞被注释为在4种参考类型中亲和力概率最高的细胞类型。sp,小穗;me,分生组织;ra,花序轴;lf,叶片。
(D)密度图显示每种细胞类型的亲和力概率分布。
(E)拟南芥花分生组织的单细胞图的UMAP图。43 通过Seurat执行降维和聚类识别。点表示单个细胞,颜色表示不同的细胞类型。在(E)和(F)中,拟南芥细胞类型包括分裂细胞(dvc)、表皮细胞(ep)、维管细胞(vs)、皮层细胞(ct)和花分生组织(fm);而水稻细胞类型包括合点(ch)、表皮(ep)、胚珠(nu)、子房(ov)、子房壁(ow)、柱头(sti)、雄蕊(sta)、簇12细胞(C12)和簇13细胞(C13)。(F)MetaNeighbor分析显示水稻雌蕊和拟南芥花分生组织细胞类型之间的相似性,用接收者操作特征曲线(AUROC)得分表示。(G)Sankey图显示水稻雌蕊细胞与水稻花序细胞(左图)或拟南芥花分生组织细胞(右图)之间的连接性。左图或右图中细胞类型之间的连接宽度分别表示细胞数量或AUROC得分。
雌蕊、花序和花序分生组织的细胞普查比较
我们基于验证的表达数据,对不同簇的细胞类型进行了进一步注释(图1D–1F、2A和2B)。以下是主要发现:
- 单一簇的空间分布:
- 簇0:代表珠心(nucellus)。
- 簇5:代表柱头(stigma)。
- 簇8:代表合点(chalaza)。
- 多个簇对应同一细胞类型:
- 表皮:簇1、2和9。
- 子房壁:簇4、6和7。
- 胚珠:簇3和10(图3A;表S2)。
尽管簇12和簇13不属于分区#1和#2,且尚未明确分配到特定细胞类型,但通过MetaNeighbor分析发现,这两个簇在两组生物学重复中表现出相似特征,分别与合点和胚珠细胞密切相关(图3B)。
(sp,小穗;me,分生组织;ra,花序轴;lf,叶片)
由于水稻雌蕊是从花序中的小穗发育而来的,我们将雌蕊的细胞普查结果与已报道的水稻发育中花序的单细胞数据集进行了比较(图3C;表S2)。主要发现如下:
- 大多数雌蕊细胞类型被注释为与小穗细胞高度相关的细胞类型,其次是主轴细胞、叶片细胞和分生组织细胞(图3C)。
- 雌蕊细胞仅与小穗细胞和主轴细胞表现出高度相似性(图3D)。
- 在关联概率大于0.95的细胞对中,大多数雌蕊细胞类型与小穗细胞相似(图3G,左图)。
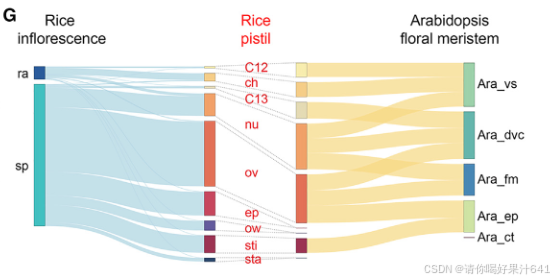
这些观察结果合理,因为雌蕊是从颖片发育而来的,而不是从花序的其他部分发育而来。种脐细胞和第12群细胞与穗轴细胞相似(图3G,左图),这表明它们在维管组织中可能具有相似的功能,如资源运输和提供机械支持。
(ch,合点;ep,表皮;nu,胚珠;ov,子房;ow,子房壁;sti,柱头;C12,簇12细胞;C13,簇13细胞)
水稻雌蕊细胞普查与拟南芥花序分生组织单细胞数据的比较
我们还将水稻雌蕊细胞普查结果与拟南芥花序分生组织的单细胞数据进行了比较(文献43)。以下是主要发现:
-
细胞类型注释
我们利用统一流形近似与投影(UMAP)绘制了拟南芥的细胞类型分布图(图3E;表S2),并通过MetaNeighbor分析发现了几种细胞类型之间的跨物种相似性。 -
水稻与拟南芥细胞类型的相似性
- 水稻的珠心细胞和胚珠细胞与拟南芥的花序分生组织细胞具有较高的相似性。
- 水稻簇13细胞与拟南芥的分裂细胞高度相似(图3F)。
(拟南芥细胞类型包括分裂细胞(dvc)、表皮细胞(ep)、维管细胞(vs)、皮层细胞(ct)和花分生组织(fm);而水稻细胞类型包括合点(ch)、表皮(ep)、胚珠(nu)、子房(ov)、子房壁(ow)、柱头(sti)、雄蕊(sta)、簇12细胞(C12)和簇13细胞(C13))

-
细胞类型的相近性
- MetaNeighbor分析表明,这些细胞类型彼此之间的相似性高于其他细胞类型,且关联可能性较高(AUROC评分 >0.6)(图3F和3G)。
- 此外,水稻的合点细胞与拟南芥的维管细胞相似,这些细胞与水稻的簇12细胞和拟南芥的皮层细胞在空间上较为接近(图3F和3G)。
结论
这些观察结果表明,不同植物物种的生殖器官在发育过程中可能保留了一些共同的细胞类型及功能特性。这种保守性为进一步研究植物发育过程中的细胞分化和功能提供了重要的线索。
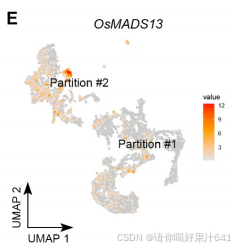
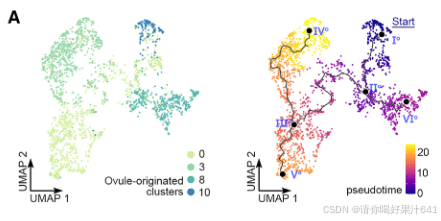
6.胚珠来源细胞的轨迹分析
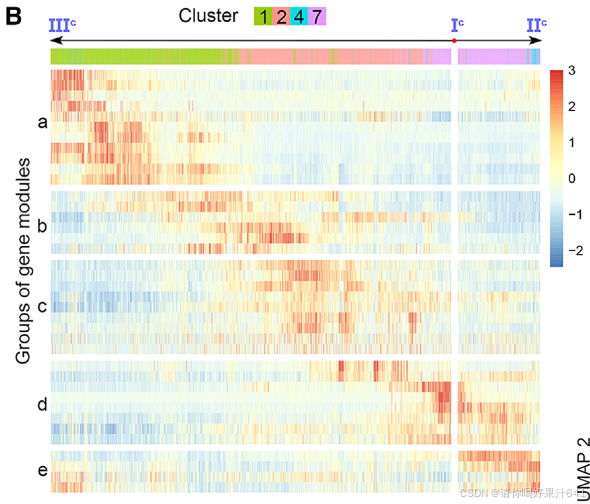
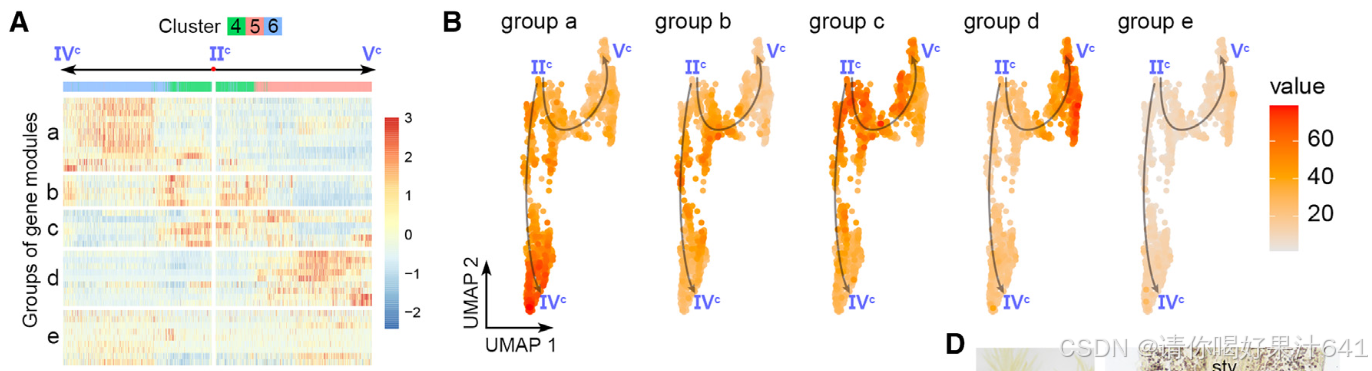
胚珠起源细胞的轨迹分析:为了评估雌蕊细胞经历的动态过程模式,我们随后对来自簇0、3、8和10的胚珠起源细胞进行了轨迹分析,这些细胞通过原位杂交和分区分析得到了确认(图2)。我们选择了表达OsMADS13最高的主要节点Io(图S5E),它在胚珠原基中高度富集,作为通过Monocle3进行伪时间分析的起点(图4A)。预测的轨迹和伪时间线包括一个“主干路径”Io–IIo–IIIo–Vo以及两条“分支路径”,IIo–VIo和IIIo–IVo(图4A)。终点主要节点VIo和IVo分别位于簇8和3(图4A),它们被注释为种脐细胞和珠被细胞(图2A-2C)。因此,分支路径指示了种脐和珠被的各自分化轨迹。分支点的伪时间意味着种脐细胞更早被指定(通过IIo),而珠被细胞稍后被指定(通过IIIo)(图4A)。这与胚珠的基端-顶端轴分化相一致。
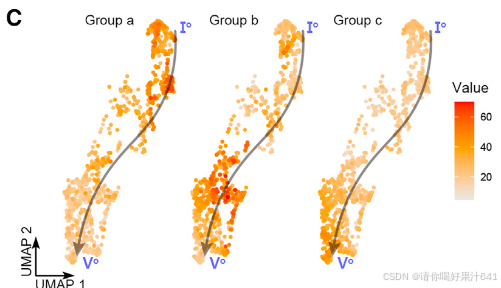
我们进一步研究了主干路径的细节。沿着伪时间动态表达的基因被整合成几个基因模块(图4B),这些基因模块进一步被聚类成三个组,即‘a’、‘b’和‘c’(图4B)。这三个组在主干路径上呈现了三波基因表达(图4B和4C),并通过基因本体(GO)富集分析揭示了它们与不同功能特征相关(图4D)。‘a’组代表了路径的起始部分,其中富集的基因与发育调控和对内源性及外源性信号的响应有关,表明它们参与调节分化程度较低的细胞对发育和环境信号的响应,而‘c’组代表了路径的末端部分,其中富集的基因与细胞分化相关的DNA复制和染色体组织有关。
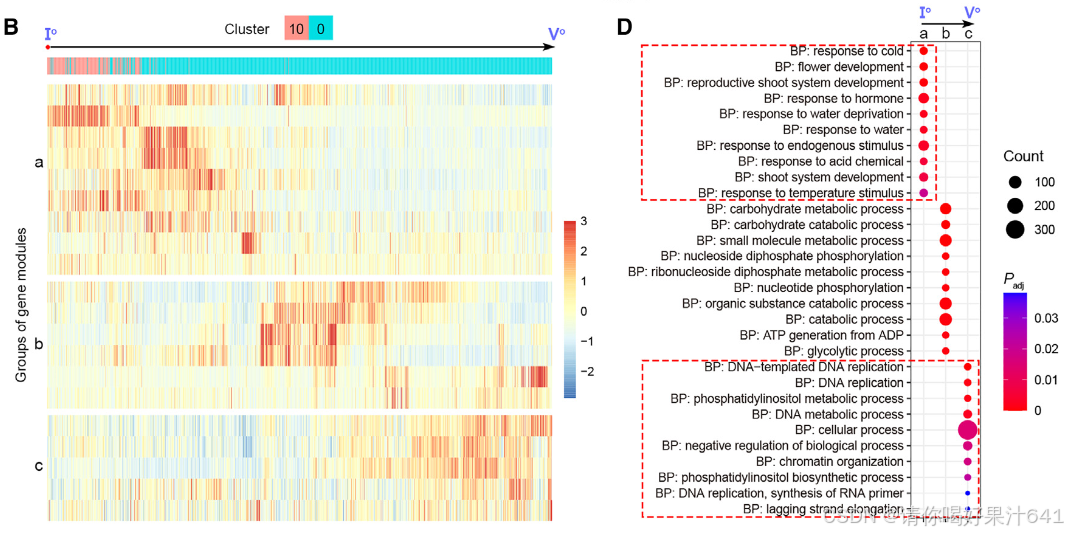
结论
轨迹分析揭示了胚珠来源细胞的动态分化模式,其中合点细胞和珠被细胞沿不同路径分化,且分化时间与胚珠基-顶轴的空间特化一致。基因表达的动态变化进一步提供了对胚珠细胞功能特性的系统理解,从未分化阶段到最终分化阶段的转变反映了胚珠发育的复杂性。
孢子体-配子体转变
我们还在通过FANS从第一批2N细胞核中分离出的假定1N细胞核上进行了snRNA-seq(图S1D-S1G),并获得了1,625个细胞核作为输入的1N细胞。有趣的是,当通过Seurat将这些细胞投影到2N细胞的UMAP结构上作为参考时,这些细胞中的大多数都与构成胚珠起源细胞主干路径的第0簇(珠心细胞)有关(图5A)。对于与第0簇有关的假定1N细胞,大多数细胞的关联概率在0.5到0.9之间,而那些关联概率超过0.95的细胞只占非常小的一部分(图5B)。对于注释为其他簇的假定1N细胞(图5A),大多数细胞的关联概率都高于0.95(图5B),这表明这些细胞是通过FANS收集的假定1N细胞核部分中2N峰值尾部的2N细胞(图S1F)。因此,我们将与第0簇有关的输入1N细胞的部分视为真正的1N细胞(在1,625个输入的1N细胞中,有989个)。
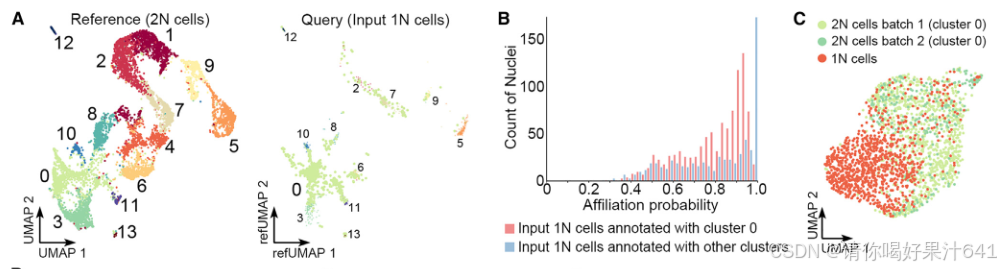
图5. 配子体-孢子体转换前的多能性重置
(A) 输入的1N细胞(查询)投影到2N细胞(参考)的UMAP结构上,由Seurat展示。颜色代表Seurat注释的聚类。每个输入的1N细胞被注释为在2N细胞的14个参考聚类中亲和力概率最高的聚类。对于输入的1N细胞,注释为簇0或其他聚类的细胞分别以点或三角形显示。(B)直方图显示预测的输入1N细胞的归属概率分布,这些细胞注释为簇0(红色)或其他聚类(蓝色)。归属概率表示细胞被注释为某个聚类的概率。此后,只有注释为簇0的输入1N细胞被视为确认的1N细胞。(C)1N细胞在簇0中与2N细胞区分开来。数据整合和UMAP可视化由Seurat执行。
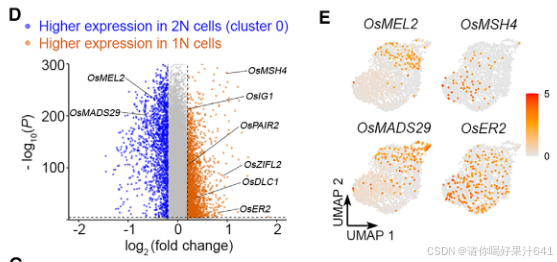
这些真正的1N细胞(989个细胞)和第0簇的2N细胞(两个批次总共1237个细胞核)通过Seurat 4.0分析被区分开来(图5C),并且表现出不同的分子特征。值得注意的是,第0簇的2N细胞和1N细胞之间的差异表达基因(DEGs)展示了孢子体-配子体转换期间的转录组轮廓变化。例如,对于减数分裂前G1/S期转换至关重要的OsMEL2和OsMADS29,或者分别负责珠心细胞降解的基因,在2N细胞中优先表达(图5D和5E;表S4)。相比之下,在1N细胞中特异性表达的包括与减数分裂相关的基因,如水稻MutS同源(MSH)家族基因OsMSH4、46、DEFECTIVE LEPTOTENE CHROMOSOME 1(OsDLC1)47和OsPAIR2、48,以及一些已知的 megasporogenesis(大孢子发生)的调控因子,如OsERECTA2(OsER2)49和水稻INDETERMINATE GAMETOPHYTE1(OsIG1)50(图5D和5E;表S4)。这些数据表明,第0簇的细胞与孢子体-配子体转换期间的减数分裂密切相关。此外,由于在胚珠起源细胞的主干路径末端富集的基因与DNA复制和染色体组织有关(图4C和4D),主要由第0簇细胞组成的主干路径代表了雌性生殖细胞的特化。
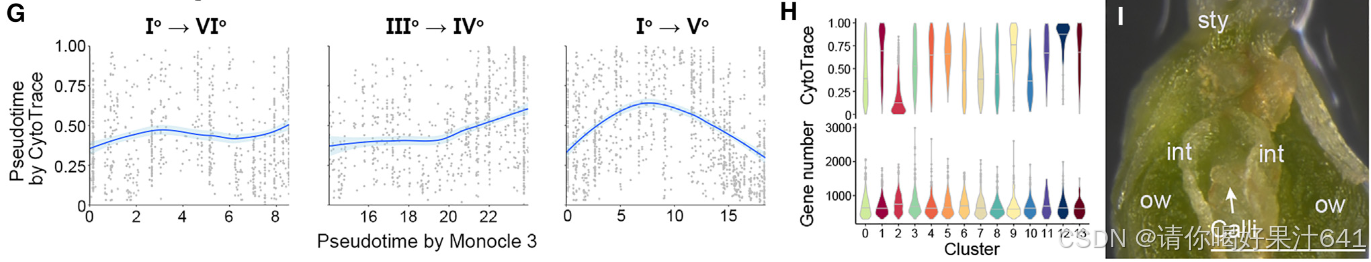
7.孢子体-配子体的转变
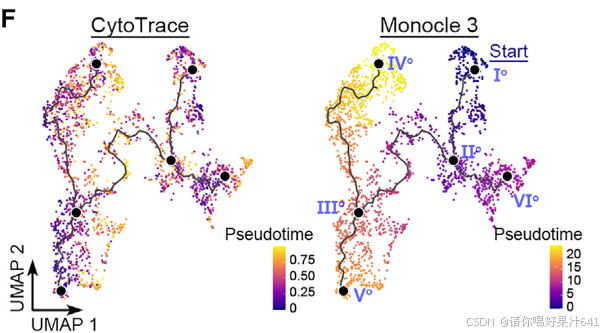
除了用于降维和图形聚类的基于图的工具Monocle3之外,我们还使用了一种非图结构的方法——利用基因计数和表达进行的细胞(cyto)轨迹重建分析(CytoTRACE),以进行轨迹分析的交叉验证。这种方法已成功用于人类细胞轨迹分析和拟南芥根发育研究,它通过检测转录多样性的减少——这在发育轨迹中的细胞伪时间投影的一个标志——来预测细胞的相对分化状态。
我们比较了由Monocle3和CytoTRACE推断的胚珠起源细胞轨迹的伪时间,发现这两种方法预测的伪时间模式在胚珠起源细胞轨迹中并不总是相关的(图5F)。它们在种脐特化路径(Io–VIo)和珠被特化后半部分(IIIo–IVo)中呈正相关,但在主干路径的后半部分(IIo–Vo)(图5G),即由珠心细胞(簇0)(图4A)构成的部分,呈负相关。由于转录多样性可能因不同簇中检测到的基因数量不同而在技术上产生误导,我们计算了不同簇的平均可检测基因数量和CytoTRACE得分。簇中检测到的平均基因数量略有变化,而CytoTRACE得分则有相当大的差异,中位数值范围从0.11到0.92(图5H)。因此,主干路径后半部分CytoTRACE值的降低不太可能是由于可检测基因数量的变化。由于从IIo–Vo的子路径描绘了向向雌性生殖细胞命运的进展,转录多样性的增加,正如主干路径后半部分CytoTRACE值的降低所揭示的,意味着在孢子体-配子体转换之前重置多能性。这一点得到了以下观察的支持:在所有细胞类型中,只有珠心细胞能够在胚珠切割后置于愈伤组织诱导培养基上时快速生成愈伤组织(图5I)。这表明珠心细胞在孢子体-配子体转变前具有较高的多能性。
表明在孢子体-配子体转变之前,细胞经历了多能性的重新获得。
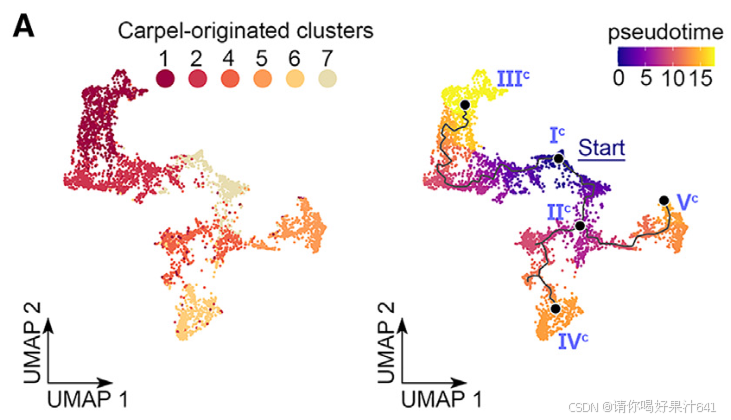
心皮来源细胞的轨迹分析
轨迹起点与路径预测
我们对心皮来源细胞进行了轨迹分析,重点评估了分区#1(图2E),但将簇9排除在外,原因如下:
- 簇9细胞核分为两部分:一部分靠近簇4,另一部分靠近簇5(图2D和2E)。
- 簇9的标记基因定位于子房壁内表面和胚珠外珠被(图2B和2C),表明簇9细胞核具有混合身份。
我们选择主节点Ic作为伪时间分析的起点,该节点的DL基因表达量最高,且DL基因在心皮原基的早期即有表达(文献7–9)。基于Monocle3,预测的轨迹从起始节点分为三条子路径(图6A):
- Ic–IIIc
- Ic–IIc–IVc
- Ic–IIc–Vc
由于Monocle3和CytoTRACE对心皮来源细胞伪时间模式的预测显示出正相关(图S7),我们主要分析了Monocle3推断的伪时间轨迹,揭示了与心皮发育相关的若干特征。
伪时间路径 Ic–IIIc:子房外表皮的特化
-
特化路径
簇1和簇2细胞具有特异性表达模式,位于子房壁外表面(图2B和2C),在轨迹路径Ic–IIIc中占主导地位(图6A),表明此路径代表子房外表皮的特化轨迹。 -
细胞表面特征
- **扫描电子显微镜(SEM)**显示,子房壁外表皮细胞紧密、规则排列,而花柱表面细胞不规则(图6E)。
- 甲苯胺蓝染色表明子房表面对染色剂具有抗性,而花柱和柱头染色性显著增强(图6F)。
- 苏丹IV染色显示子房壁外表皮的角质层完整,而花柱的角质层部分受损,柱头完全缺失(图6G)。
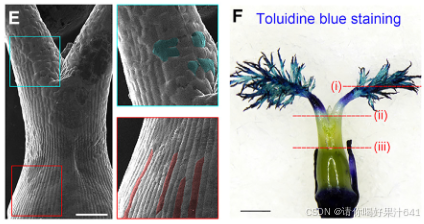
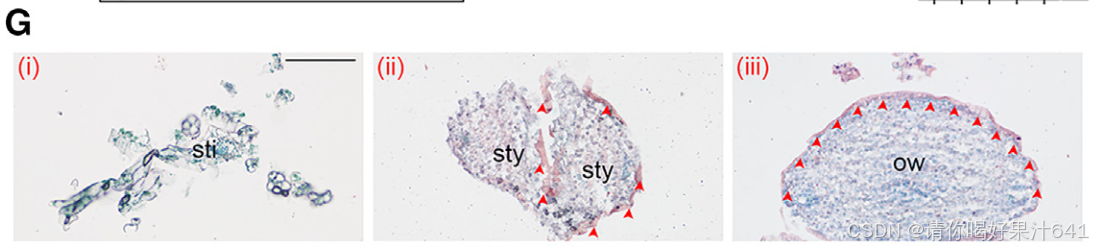
这些结果表明,伪时间路径Ic–IIIc专门描述了子房外表皮的特化过程。
伪时间路径 IIc–IVc 和 IIc–Vc 的功能分化
-
路径 IIc–IVc:花柱基部发育轨迹
- 基因模块的表达动力学分析显示,基因分为四组(“a”到“d”)(图7A)。
- 组“a”基因主要在簇6细胞中表达,富集于终点节点IVc,与光合作用相关功能相关(图7A–7C)。
- I2-KI染色在花柱基部检测到富含淀粉的区域,表明活跃的光合作用(图7E)。这与簇6标记基因在同一区域的表达一致(图2B),表明路径IIc–IVc描述了花柱基部发育的轨迹。
-
路径 IIc–Vc:柱头命运的分化轨迹
- 组“d”基因在簇5柱头细胞中表达(图S3C),富集于终点节点Vc,并与柱头相关GO术语(如脂质代谢)显著相关(图7A–7C)。
- 路径IIc–Vc描述了柱头命运的分化轨迹。
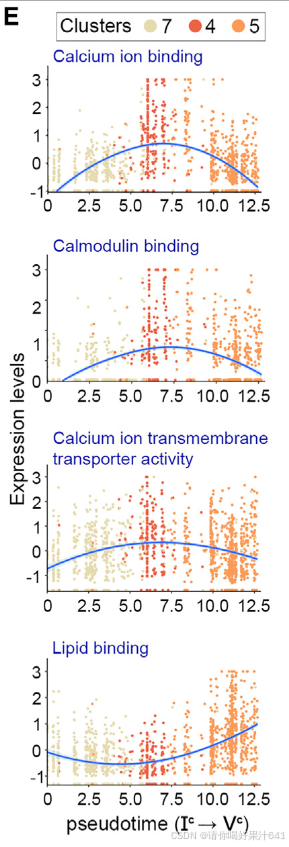
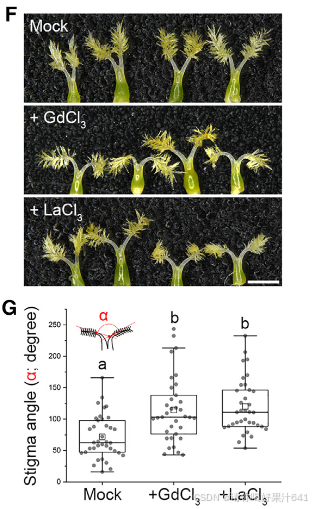
第三,'b’组和’c’组基因在分支点IIc处富集,与钙离子结合(分子功能[MF])、钙调蛋白结合(MF)、钙离子跨膜运输活动(MF)、信号传导(生物过程[BP])和细胞通讯(BP)等功能相关(图7A-7C)。在整个柱头发育轨迹(Ic–IIc–IVc)的伪时间线上,这些与钙相关的基因在中期特定表达,而’d’组基因在柱头细胞中的表达仅出现在过程的末期(图7E),这表明钙相关信号在柱头特化之前的风格发育中起着作用。然后,我们应用了成熟的钙通道阻断剂LaCl3和GdCl3来处理开花前一天的幼嫩雌蕊,这明显增大了次日柱头之间的角度(图7F和7G)。这支持了钙信号在柱头发育的各个方面,如其伸展,的参与。
总结
轨迹分析揭示了心皮来源细胞的分化路径,其中:
- Ic–IIIc路径特化为子房外表皮。
- IIc–IVc路径描述了花柱基部发育。
- IIc–Vc路径指向柱头分化命运。
钙信号在柱头发育过程中发挥重要作用,特别是在柱头分化早期的信号传导和细胞命运特化中起关键调控作用。
讨论
尽管在过去的几十年中,许多研究致力于了解植物的生殖发育,但对于被子植物中承载孢子体和配子体世代关键转变的雌蕊,其细胞分化的系统性视角仍然未知。在水稻中,基于从小花和花序分生组织中制备的原生质体的单细胞测序研究(文献42)已经揭示了早期花序和小花发育的分化轨迹,但雌蕊中发育的各种细胞类型如何发挥其独特而多样化的作用仍不清楚。这些作用包括生成雌配子、提供利于受精的环境以及保护种子发育。
为了解决这些关键知识空白,本研究构建了一个全面的时空雌蕊单核RNA测序(snRNA-seq)图谱,并揭示了在单细胞水平上胚珠和心皮来源细胞轨迹的细胞组成。通过建立改良的基于液滴的snRNA-seq方法以及ab initio标记鉴定和细胞类型注释,我们对水稻雌蕊中的细胞进行了生物学注释。值得注意的是,每种细胞类型都与特定的基因模块相关,这些基因模块表明了这些细胞类型在雌蕊中的基本作用(图S6A)。例如:
- 表皮细胞富含高度表达于细胞外围并与化学刺激和损伤响应相关的基因(模块b)(图S6A;表S3)。
- 子房壁(包括花柱基部)中的基因主要参与钙信号和细胞通信(模块e)(图S6A;表S3),这些过程对于柱头的展开至关重要(图6G和7F)。
- 胚珠和珠心细胞中的基因高度参与RNA处理和细胞分裂(模块a)(图S6A;表S3)。
- 珠心细胞相比其他胚珠细胞,更富含与ATP生成相关的基因(模块f)(图S6A;表S3),这表明能量代谢在配子发育及其准备受精中的重要性。
时空信息的整合
从开花前1天的穗中收集未受精雌蕊,还使我们能够研究雌蕊在发育过程中发生的连续变化(图1A和1B)。由于水稻小穗以基部向上的顺序开花,我们的细胞普查除了空间信息外,还包括水稻雌蕊发育轨迹的时间信息(图S6B)。例如:
- 簇10和簇7分别代表了年轻胚珠和未分化心皮细胞类型,这些细胞中花器官身份基因高度表达。
- 其他簇代表了完全发育的胚珠(簇0、3和8)或心皮(簇1、2、4、5、6和9)的细胞类型。
胚珠和心皮发育的差异
-
胚珠发育:
- 与胚珠和雌蕊发育相关的基因在年轻胚珠和心皮细胞中均有富集。
- 然而,与RNA处理和染色体组织相关的基因仅在年轻和发育完成的胚珠细胞中高度表达(图S6B;表S3),这表明胚珠维持活跃的核过程以支持雌配子发育的持续轨迹。
-
心皮发育:
- 在发育完成的心皮细胞中高度表达的基因未与任何发育完成的胚珠富集的GO术语相关(图S7B)。
- 这表明,尽管胚珠和心皮在雌蕊中同时发育,但它们在主要生物过程和潜在分子机制上有很大差异。
研究意义
本研究通过改良的液滴式snRNA-seq方法,结合原位杂交、批量RNA测序和轨迹分析,提供了一种集成手段来研究水稻雌蕊的细胞普查。由于雌蕊对原生质体制备的抗性和缺乏发育中雌蕊的有效细胞标记,这些特性难以通过scRNA-seq研究。本研究中发现的全面时空snRNA-seq细胞普查使我们能够在单细胞水平上解析水稻雌蕊的细胞异质性和发育轨迹的调控事件,为更好地理解植物雌性生殖发育及其基因调控网络提供了一步重要进展。
展望
总之,我们的细胞普查为深入研究组成整个雌蕊的各种细胞类型提供了基础,这些细胞需要在胚珠和心皮来源细胞中实现雌配子发育和受精准备的无缝调控。将RNA测序与快速发展的单细胞多组学技术结合,有望全面阐明植物生殖发育背后的细胞特征,并推动与作物改良相关的技术创新。
研究的局限性
我们的单核RNA测序(snRNA-seq)样本仅包括了开花前1天采集的水稻穗。这种采样方法虽然能够研究在开花前依次发育的雌蕊,但若要推断整个雌蕊发育过程中细胞分化的轨迹分析,还需要更广泛的时间窗口进行采样。
此外,尽管我们区分了1N细胞和2N细胞,但由于捕获到的细胞数量较少且异质性有限,分辨率可能不足。针对1N细胞开发并应用特定的富集策略可能会有所帮助,这将有助于更深入地研究这些细胞及其与其他雌蕊发育相关细胞的关系。
STAR+方法
在线版论文中提供了详细的研究方法,内容包括:
实验模型和研究对象细节
- 水稻
方法细节
- 通过荧光激活核分选(FANS)进行细胞核分离
- 初步核计数
- 原位杂交
- 组织化学分析
- LaCl₃和GdCl₃处理
定量和统计分析
- 原始snRNA-seq数据的预处理
- 双细胞检测和数据过滤
- 批次整合
- 聚类与簇特异性标记检测
- 细胞普查的比较
- 轨迹和子路径分析
- 伪时间分析
- 数据整合与投影
- 轨迹中的动态表达基因分析
- 基因本体(GO)与通路富集分析
- 批量RNA测序数据分析
补充信息
补充信息可在线访问:
https://doi.org/10.1016/j.
STAR+方法关键资源表
试剂或资源
| 类别 | 来源 | 标识符 |
|---|---|---|
| 抗体 | ||
| 抗地高辛-AP Fab片段 | Roche | Cat#11093274910;RRID: AB_2734716 |
| 化学品、肽和重组蛋白 | ||
| 苏丹IV | Sigma-Aldrich | Cat# 198102 |
| 甲苯胺蓝 | Sigma-Aldrich | Cat# 89640 |
| 碘化丙啶 | Sigma-Aldrich | Cat# P4170 |
| RNaseOUT | Invitrogen | Cat# 10777019 |
| Protector RNase 抑制剂 | Merck | Cat# 3335399001 |
| 氯化镧 (LaCl₃) | Sigma-Aldrich | Cat# 203521 |
| 氯化钆 (GdCl₃) | Sigma-Aldrich | Cat# G7532 |
| NBT/BCIP储备溶液 | Roche | Cat# 11681451001 |
| RNA聚合酶SP6 | Roche | Cat# 11487671001 |
关键商用试剂盒
| 试剂盒 | 来源 | 标识符 |
|---|---|---|
| Chromium Next GEM 单细胞3’试剂盒 v3.1 | 10x Genomics | Cat# 1000269 |
| Chromium Next GEM 单细胞3’ LT v3.1 | 10x Genomics | Cat# 1000325 |
| 双重索引试剂盒 TT Set A | 10x Genomics | Cat# 1000215 |
| Chromium Next GEM 芯片 G 单细胞试剂盒 | 10x Genomics | Cat# 1000127 |
| Chromium Next GEM 芯片 L 单细胞试剂盒 | 10x Genomics | Cat# 1000321 |
| RNeasy Micro Kit | Qiagen | Cat# 74004 |
数据存储
| 资源 | 来源 | 标识符 |
|---|---|---|
| snRNA-seq实验数据 | 本研究 | GEO (GSE208433) |
| 实验中使用的引物,请参考表S5 | 本研究 | N/A |
软件与算法
资源可用性
主要联系人
如需进一步的信息或资源和试剂的请求,请联系主要联系人:Hao Yu(邮箱:dbsyuhao@nus.edu.sg)。
材料可用性
本研究未生成新的独特试剂。
数据和代码的可用性
- snRNA-seq数据已上传至GEO数据库(GSE208433),自发表之日起公开可用。
- 数据处理代码可在GitHub获取:https://github.com/yuhaolab/snrnaseq。
- 任何其他需要重新分析本研究数据的信息,可通过联系主要联系人获得。
实验模型和研究对象细节
水稻
- 水稻(Oryza sativa ssp. Japonica cultivar Nipponbare)植株种植在混合沙土的盆栽土壤中。
- 生长环境为气候室,相对湿度70%,昼夜节律如下:
- 10小时光照,温度30°C;
- 14小时黑暗,温度25°C。
方法细节
通过荧光激活核分选(FANS)进行细胞核分离
流式细胞术的倍性分析基于改良的前人协议。
- 将解剖后的雌蕊(每管15个,共两管)放入1.5 mL的安全锁EP管中,在液氮中冷冻并研磨成粉末。
- 随后加入500 µL细胞核分离缓冲液(1x DPBS,0.04% BSA,2 mM MgCl₂,0.015% NP-40,0.015% Tween-20,8 U/mL RNaseOUT,4 U/mL Protector RNase抑制剂)悬浮组织颗粒。
- 用10 µm的微型细胞筛过滤后,以500 rcf离心5分钟(4°C)。
- 移除上清液后,将沉淀重悬于250 µL重悬缓冲液(1x DPBS,0.04% BSA,8 U/mL Protector RNase抑制剂)。
- 加入25 µL碘化丙啶(PI)储液(1 mg/mL,经过0.22 µm滤膜过滤的无RNase水),使细胞核粗悬浮液保持冰上冷却。
- 在4°C下进行FANS操作,并用200 µL重悬缓冲液清洗收集管(1.5 mL安全锁EP管),同时作为着陆缓冲液。收集管中总量达到500 µL时更换管。
- 分选出的细胞核以500 rcf离心5分钟(4°C)。移除上清液后,将细胞核重悬于60 µL重悬缓冲液中。
- 根据初步实验调整的细胞核计数,2N细胞核(约10,000个)和1N细胞核(约3,000个)分别加载到微流控芯片(10x Genomics)上,使用Chromium Next GEM Single Cell 3’ Kit v3.1和Chromium Next GEM Single Cell 3’ LT v3.1进行GEM分区。
初步细胞核计数
- 使用完整的小穗(4个)进行FANS辅助的细胞核分离(如上所述)。
- 将1 µL细胞核悬浮液分装到15孔滑片(ibidi,Cat. No: 81506)的每个孔中,并用EVOS M7000成像系统(Thermo Fisher Scientific)捕捉堆积模式图像。
- 用ImageJ和EVOS M7000内置功能进行基于图像的细胞核计数,并与分选仪记录的计数结果进行比较。
- 独立样本的多次重复实验显示分选仪记录计数与图像验证计数的稳定比率。根据该比率调整分选仪计数,以处理单核文库构建样品。
原位杂交
- 扩增的候选标记基因特定区域分别在其扩增产物的5’或3’末端融合T7或SP6启动子序列。
- 使用DIG RNA标记试剂盒(Roche Molecular Biochemicals)进行体外转录。
- 石蜡切片的原位杂交和整体原位杂交实验按照之前报道的方法进行。
组织化学分析
-
石蜡切片的I2-KI染色和苏丹IV染色与原位杂交的制备方式类似。
- I2-KI染色液(3.3 g/L I2,6.7 g/L KI水溶液)用于切片。染色1分钟后,切片快速用水洗涤并用50%甘油封片,进行显微镜观察。
- 苏丹染色液(50 mg/mL苏丹IV溶于二甲基亚砜)用于切片,在70°C下染色15分钟。染色后,切片用70%乙醇清洗两次,再用0.1%甲苯胺蓝染色2分钟,最后用水洗涤并用50%甘油封片。
-
对整体I2-KI染色:
- 固定的雌蕊先用甲醇去除叶绿素5分钟,再在水中重水化10分钟,转移至I2-KI染色液中,在室温下染色30分钟。
- 染色后用水清洗两次,并用氯化水合溶液(8 g氯化水合,2 mL甘油,4 mL H₂O)封片,使用体视显微镜(Nikon, SMZ18)观察。
LaCl₃ 和 GdCl₃ 处理
- 从开花前一天的穗轴上解剖出小穗,去除稃片后,将开放的小穗固定在6孔板底部,使雌蕊朝上。
- 用以下溶液喷洒雌蕊:
- LaCl₃(500 mM),
- GdCl₃(500 mM),
- KCl(500 mM,作为模拟处理),
溶解于含0.02% Silwet L-77的10 mM Tris(pH 6.5)中。
- 封闭6孔板,在25°C下孵育24小时后,用体视显微镜(Nikon, SMZ18)观察。
- 使用ImageJ测量柱头之间的夹角。
定量和统计分析
原始snRNA-seq数据预处理
- 原始snRNA-seq FASTQ文件使用Cell Ranger 6.1.2(10x Genomics)进行预处理。
- 首先将读取的序列比对至水稻基因组(IRGSP-1.0),基因组和GTF注释文件从Ensembl Plants(版本52)下载(https://plants.ensembl.org)。
- 使用
cellranger mkref功能筛选GTF注释,并构建包含以下基因类型的参考文件:protein_coding(蛋白编码基因),antisense_RNA(反义RNA),nontranslating_CDS(非翻译CDS),pre_miRNA(前体miRNA),rRNA(核糖体RNA),sense_intronic(正义内含子),snoRNA(小核仁RNA),snRNA(小核RNA)。
- 使用
cellranger count生成单细胞基因计数矩阵,并设置参数--include-introns,以包括比对至基因内含子区域的读取序列。 - 样本中超过95%的读取序列通过Cell Ranger内置的STAR比对器成功比对至IRGSP-1.0参考基因组。
- Cell Ranger生成的基因-细胞计数矩阵作为后续分析的输入数据。
双细胞检测和数据过滤
- 使用**DoubletFinder(版本2.0.3)**检测每个snRNA-seq数据集中的双细胞(doublet)。标记为“Doublet”的细胞被移除。
- 为进一步过滤低质量的细胞和基因,使用**Seurat(版本4.0.4)**读取并处理基因-细胞矩阵:
- 保留在至少5个细胞中表达的基因。
- 保留表达基因数量介于300到3000之间的细胞。
- 移除表达线粒体基因占比超过10%或表达叶绿体基因占比超过5%的细胞。
- 随后的分析基于过滤后的基因-细胞矩阵进行。
批次整合
- 对两个批次的snRNA-seq数据集进行数据过滤后,进一步基于细胞的UMI计数分布过滤批次2的数据:
- 比较批次1和批次2细胞的UMI计数分布。
- 移除批次2中UMI计数低于批次1中90%细胞UMI计数的细胞(见图S2B)。
- 最终,批次1保留6,004个细胞,批次2保留2,169个细胞,使用Seurat进行整合。
- 使用
FindIntegrationAnchors函数(reference参数设置为批次1)检测整合锚点,并基于这些锚点使用IntegrateData函数整合批次1和批次2的表达矩阵。后续分析基于整合后的基因-细胞矩阵进行。
聚类和特定簇标记基因检测
- 使用Seurat进行聚类和簇特异性标记基因检测:
- 对整合的Seurat对象进行缩放,并基于批次1细胞的前2000个可变特征构建所有细胞的主成分分析(PCA)。
- 使用
FindNeighbors函数(降维数为60)和FindClusters函数(分辨率为0.4)识别簇。 - 使用
FindAllMarkers函数检测簇特异性基因标记,采用默认的Wilcoxon秩和检验,显著标记的选择标准为:- P值 < 0.01,
- 平均对数2倍数变化(log2 fold change) > 1,
- 表达基因的细胞比例(ptc) > 0.2。
- 使用**UMAP(统一流形近似与投影)**降维可视化簇结构。
- 表S1和S2中标记基因的注释基于**The Rice Annotation Project(RAP, https://rapdb.dna.affrc.go.jp/)**完成。
细胞普查的比较
细胞普查比较
细胞普查的比较通过MetaNeighbor工具进行。相似性基于MetaNeighbor预测的AUC(曲线下面积)得分来衡量,AUC得分越高表示细胞类型或批次之间的相似性越高。
-
数据来源:
- 拟南芥花原基的snRNA-seq数据从GEO数据库下载(GSE174599)。
使用的是处理过的第5阶段花的基因-细胞矩阵,用于比较水稻雌蕊和拟南芥花原基之间的细胞类型。 - 水稻和拟南芥基因的同源基因表从Ensembl Plants(版本52)下载,并用来结合拟南芥和水稻的基因ID。
- 水稻小花和花序分生组织的scRNA-seq数据从GEO数据库下载(GSE185068)。
使用处理过的5个样本的基因-细胞矩阵作为参考,用于比较水稻雌蕊和花序之间的细胞类型。
- 拟南芥花原基的snRNA-seq数据从GEO数据库下载(GSE174599)。
-
分析方法:
- 拟南芥和水稻数据集作为不同的研究对象处理,使用“variableGenes”选择顶部变量基因特征。
- 使用“MetaNeighborUS”计算拟南芥和水稻不同细胞类型之间的AUC分数。
- 水稻雌蕊的整合Seurat对象作为查询样本,通过“MapQuery”函数投射到花序参考样本中。
- 此过程整合了参考样本和查询样本,并生成每个查询细胞归属于参考样本中注释的细胞类型的关联概率。
轨迹和子路径分析
-
轨迹分析工具:
- 使用Monocle3(版本1.0.0)进行轨迹分析。
- 基因-细胞矩阵从Seurat对象转换为Monocle3的“cell_data_set”对象,转换使用SeuratWrappers R包。
- 使用“align_cds”函数纠正批次效应。
-
分析过程:
- 轨迹分析按以下步骤执行:
- 使用“preprocess_cds”和“align_cds”函数对数据进行归一化和预处理,为轨迹推断做好准备。
- 使用非线性降维方法(UMAP),通过“reduce_dimension”函数将数据投射到低维空间。
- 使用“cluster_cells”函数将细胞聚类到不同的分区中。
- 使用“learn_graph”函数学习每个分区的轨迹。
- 通过“plot_cells”函数可视化细胞和轨迹。
- 轨迹分析按以下步骤执行:
-
轨迹和分区特异性:
- 分区特异性轨迹的生成和可视化按照与上述相同的过程由Monocle3完成。
伪时间分析
-
伪时间推断工具:
- 使用Monocle3和CytoTRACE进行伪时间推断。
-
伪时间起始节点:
- 对每组细胞,轨迹的起始节点根据Seurat簇注释确定为较少分化的组织位置。
- 根据细胞到基节点沿轨迹的距离,使用Monocle3估算其他细胞的伪时间。
-
CytoTRACE分析:
- CytoTRACE自动基于细胞中表达基因的数量,估算每个细胞的分化伪时间。
- CytoTRACE推断的伪时间范围为0到1,其中0代表更多分化的状态,1代表较少分化的状态。
- 为便于与Monocle3推断的伪时间进行比较,将CytoTRACE推断的伪时间转换为1-伪时间。
数据整合与投射
- 样本整合:
重复样本和推测的1N样本通过Seurat分别与已分析样本(2N,批次1)整合。
这些样本作为查询样本,利用MapQuery函数投射到参考样本(本研究中分析的样本)中。
MapQuery函数整合了参考样本与查询样本,并生成每个查询细胞属于参考样本中注释细胞类型的概率。
轨迹中动态表达基因的识别
-
子路径基因识别:
使用Monocle3识别每个子路径中的动态表达基因。
对于特定主节点之间的每个子路径,轨迹中的动态表达基因由graph_test函数识别,P值小于0.05的基因被进一步通过find_gene_modules聚类到基因模块中。
每个模块中的基因表达通过aggregate_gene_expression函数进行整合。 -
模块聚类与可视化:
对单一子路径或分支子路径,基因模块基于细胞中的表达通过层次聚类分析进一步聚类。
“空间”和“时间”模型(图S6)包括Seurat识别的簇0-10中的细胞。- 轨迹首先通过UMAP使用learn_graph函数学习。
- 轨迹中的动态表达基因通过graph_test函数识别。
- 为选择特定类型的基因模块(根据空间或时间信息的特定细胞类型),选择在某特定细胞类型中表达超过10%的基因(P值 < 0.05),并通过find_gene_modules聚类为基因模块。
- 每个细胞类型的模块表达由aggregate_gene_expression函数整合,模块表达分数通过圆形条图可视化。
基因本体(GO)和通路富集分析
- 富集工具:GO和KEGG通路富集通过gprofiler2(版本0.2.1)执行。
- 显著性筛选:
- 显著富集功能的选择条件为P值小于0.05。
- 按P值从小到大排序,选择最显著的富集术语。
批量RNA-Seq数据分析
-
数据对齐:批量RNA-Seq样本使用STAR对齐至IRGSP-1.0参考基因组。
-
基因计数:使用featureCounts生成基因的reads计数。
-
伪批量表达:
伪批量snRNA-seq基因表达通过将所有细胞中基因表达值相加生成。
伪批量基因表达与批量基因表达之间的相关性通过Spearman等级相关系数测量。 -
差异表达基因(DEG)分析:
使用DEseq2识别雌蕊上部与下部之间的差异表达基因(DEGs)。
筛选条件:调整后的P值小于0.05且|log2(倍数变化)|大于1的基因。


















 7151
7151

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









