







 超级会员免费看
超级会员免费看
1. 软件安装
示例数据下载
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("karyoploteR")
BiocManager::install("BSgenome.Hsapiens.UCSC.hg19")
BiocManager::install("regioneR", force = TRUE)
2. LRR - BAF与CN关系
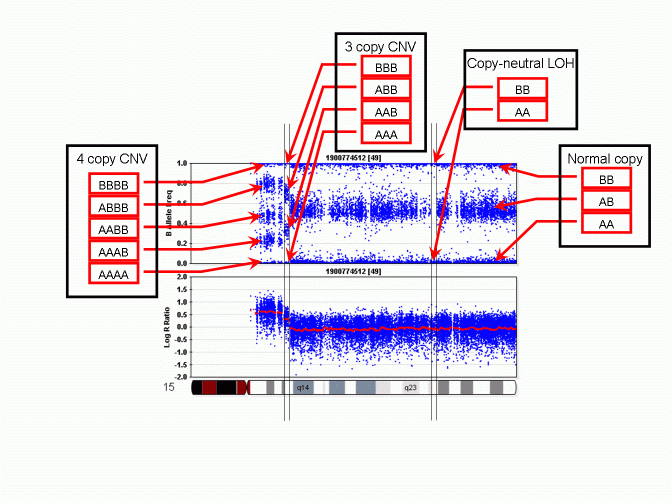
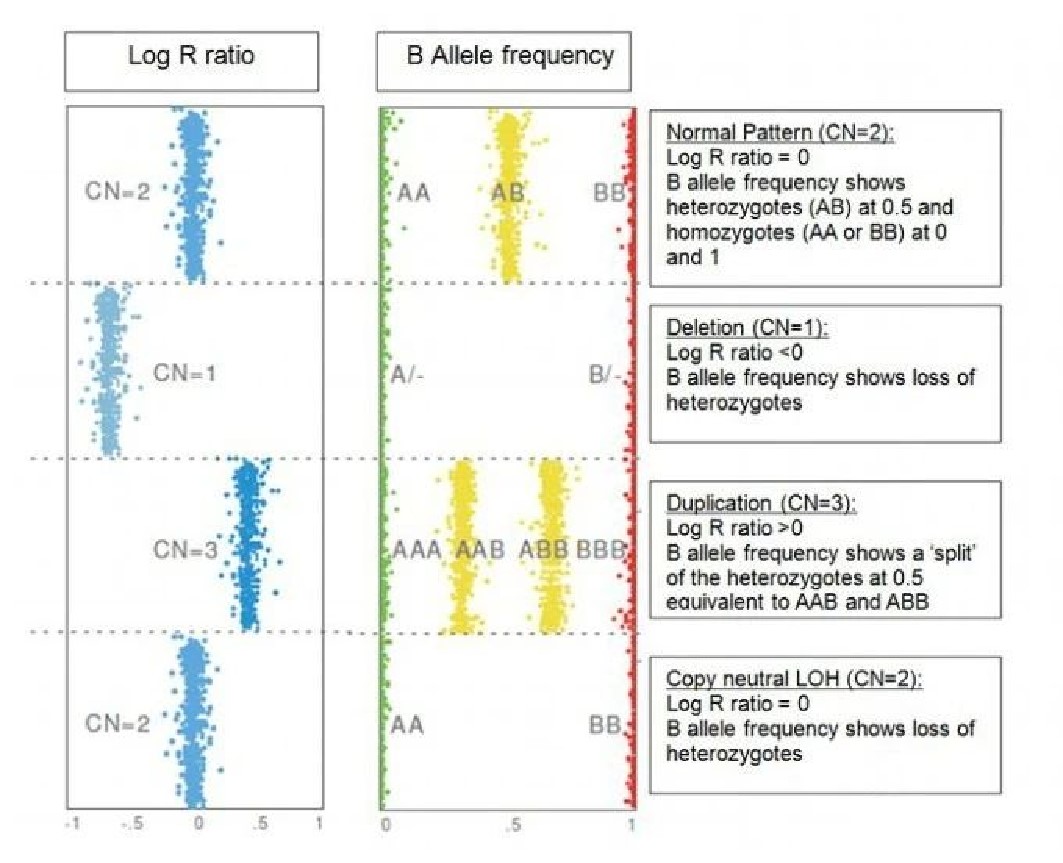
3. SNP芯片CNV分析可视化
SNPArray.26T.txt.gz示例数据文件可在本人github下载:
https://github.com/LittleGlowRobot/Jackwu/blob/main/data/SNPArray.26T.txt.gz
保存至snps_array文件夹。
# 设置工作目录
setwd("D:/snps_array")
dir()
library(karyoploteR)
genome.version <- 'hg19'
# 示例数据
data.file <- "SNPArray.26T.txt.gz"
snp.data <- read.table(file = data.file, sep = '\t',
header = TRUE, stringsAsFactors = FALSE)
head(snp.data)
# SNP.Name Sample.ID Allele1...Top Allele2...Top GC.Score Sample.Name Sample.Group
# 1 rs11152336 34 A C 0.7622 26T 26T
# 2 rs4458740 34 A C 0.8454 26T 26T
# 3 rs4264565 34 C C 0.8317 26T 26T
# 4 rs4474684 34 G G 0.7729 26T 26T
# 5 rs7677662 34 A A 0.7739 26T 26T
# 6 rs4682434 34 A C 0.4395 26T 26T
# Sample.Index SNP.Index SNP.Aux Allele1...Forward Allele2...Forward Allele1...Design
# 1 32 81073 0 A C A
# 2 32 443708 0 A C T
# 3 32 434421 0 C C G
# 4 32 444477 0 C C C
# 5 32 613842 0 A A A
# 6 32 456548 0 T G A
# Allele2...Design Allele1...AB Allele2...AB Allele1...Plus Allele2...Plus Chr Position
# 1 C A B A C 18 59864223
# 2 G A B A C 7 156089537
# 3 G B B C C 2 215283259
# 4 C B B C C 16 951045
# 5 A A A A A 4 145513053
# 6 C A B T G 3 112534069
# GT.Score Cluster.Sep SNP ILMN.Strand Customer.Strand Top.Genomic.Sequence
# 1 0.7679 0.7687 [A/C] TOP TOP NA
# 2 0.8203 0.7304 [T/G] BOT TOP NA
# 3 0.8107 1.0000 [T/G] BOT TOP NA
# 4 0.7734 0.9690 [T/C] BOT BOT NA
# 5 0.7741 0.9954 [A/G] TOP TOP NA
# 6 0.7903 0.6738 [A/C] TOP BOT NA
# Plus.Minus.Strand Theta R X Y X.Raw Y.Raw B.Allele.Freq Log.R.Ratio
# 1 + 0.651 0.642 0.243 0.398 1266 1753 0.6157 -0.3307
# 2 - 0.615 1.137 0.465 0.672 2350 2903 0.4573 0.1754
# 3 - 0.972 1.266 0.053 1.213 486 5697 1.0000 -0.0784
# 4 + 0.974 1.161 0.046 1.115 432 4710 1.0000 -0.1139
# 5 + 0.033 1.369 1.300 0.068 6213 460 0.0000 -0.0547
# 6 - 0.330 0.465 0.296 0.169 1744 866 0.3379 -0.1655
# CNV.Value CNV.Confidence GC_probe GCC_LRR
# 1 NA NA 0.31 -0.26
# 2 NA NA 0.44 0.18
# 3 NA NA 0.31 -0.01
# 4 NA NA 0.62 -0.15
# 5 NA NA 0.36 -0.01
# 6 NA NA 0.30 -0.10
colnames(snp.data)
# [1] "SNP.Name" "Sample.ID" "Allele1...Top"
# [4] "Allele2...Top" "GC.Score" "Sample.Name"
# [7] "Sample.Group" "Sample.Index" "SNP.Index"
# [10] "SNP.Aux" "Allele1...Forward" "Allele2...Forward"
# [13] "Allele1...Design" "Allele2...Design" "Allele1...AB"
# [16] "Allele2...AB" "Allele1...Plus" "Allele2...Plus"
# [19] "Chr" "Position" "GT.Score"
# [22] "Cluster.Sep" "SNP" "ILMN.Strand"
# [25] "Customer.Strand" "Top.Genomic.Sequence" "Plus.Minus.Strand"
# [28] "Theta" "R" "X"
# [31] "Y" "X.Raw" "Y.Raw"
# [34] "B.Allele.Freq" "Log.R.Ratio" "CNV.Value"
# [37] "CNV.Confidence" "GC_probe" "GCC_LRR"
snp.GRanges <- regioneR::toGRanges(snp.data[, c("Chr", "Position", "Position",
"B.Allele.Freq", "Log.R.Ratio")])
snp.GRanges
# GRanges object with 100000 ranges and 2 metadata columns:
# seqnames ranges strand | B.Allele.Freq Log.R.Ratio
# <Rle> <IRanges> <Rle> | <numeric> <numeric>
# 1 18 59864223 * | 0.6157 -0.3307
# 2 7 156089537 * | 0.4573 0.1754
# 3 2 215283259 * | 1.0000 -0.0784
# 4 16 951045 * | 1.0000 -0.1139
# 5 4 145513053 * | 0.0000 -0.0547
# ... ... ... ... . ... ...
# 99996 2 141426726 * | 0.0115 -0.2476
# 99997 2 215052524 * | 0.0031 -0.0609
# 99998 2 137508544 * | 1.0000 -0.1405
# 99999 2 868450 * | 0.0000 -0.1733
# 100000 22 25618890 * | 0.6797 -0.1298
# -------
# seqinfo: 26 sequences from an unspecified genome; no seqlengths
# 修改snp.GRanges右侧metadata列名
names(mcols(snp.GRanges)) <- c("BAF", "LRR")
seqlevelsStyle(snp.GRanges) <- "UCSC"
head(snp.GRanges)
# GRanges object with 6 ranges and 2 metadata columns:
# seqnames ranges strand | BAF LRR
# <Rle> <IRanges> <Rle> | <numeric> <numeric>
# 1 chr18 59864223 * | 0.6157 -0.3307
# 2 chr7 156089537 * | 0.4573 0.1754
# 3 chr2 215283259 * | 1.0000 -0.0784
# 4 chr16 951045 * | 1.0000 -0.1139
# 5 chr4 145513053 * | 0.0000 -0.0547
# 6 chr3 112534069 * | 0.3379 -0.1655
# 提取LRR小于-3的索引
lrr.below <- which(snp.GRanges$LRR < -3)
# 根据索引修改LRR值为-3
snp.GRanges$LRR[lrr.below] <- -3
library(biomaRt)
# genes.symbol <- c("PKD1","BRCA1","BRCA2")
genes.symbol <- c("PKD1")
# 查看支持版本
# listEnsemblArchives()
ensembl <- useEnsembl(biomart = "ensembl", dataset = "hsapiens_gene_ensembl",
version = 108)
genes <- regioneR::toGRanges(getBM(attributes = c('chromosome_name', 'start_position', 'end_position', 'hgnc_symbol'),
filters = 'hgnc_symbol', values = genes.symbol, mart = ensembl))
genes
# GRanges object with 3 ranges and 1 metadata column:
# seqnames ranges strand | hgnc_symbol
# <Rle> <IRanges> <Rle> | <character>
# 1 17 43044295-43170245 * | BRCA1
# 2 13 32315086-32400268 * | BRCA2
# 3 16 2088708-2135898 * | PKD1
# -------
# seqinfo: 3 sequences from an unspecified genome; no seqlengths
seqlevelsStyle(genes) <- "UCSC"
pp <- getDefaultPlotParams(plot.type = 4)
pp$data1inmargin <- 2
# 将cytoband转换为GRanges对象
# chr1 0 2300000 p36.33 gneg
# chr1 2300000 5400000 p36.32 gpos25
# chr1 5400000 7200000 p36.31 gneg
cytoband.data <- read.table("cytoBand.txt", sep = "\t",
header = FALSE, stringsAsFactors = FALSE)
# 修改列名
colnames(cytoband.data) <- c("chrom", "chromStart", "chromEnd", "name", "gieStain")
head(cytoband.data)
cytoband.GRanges <- regioneR::toGRanges(cytoband.data[, c("chrom", "chromStart", "chromEnd", "name", "gieStain")])
cytoband.GRanges
# GRanges object with 862 ranges and 2 metadata columns:
# seqnames ranges strand | name gieStain
# <Rle> <IRanges> <Rle> | <character> <character>
# 1 chr1 0-2300000 * | p36.33 gneg
# 2 chr1 2300000-5400000 * | p36.32 gpos25
# 3 chr1 5400000-7200000 * | p36.31 gneg
# 4 chr1 7200000-9200000 * | p36.23 gpos25
# chrom.all <- c(paste0("chr", seq(1, 22, 1)), "chrX", "chrY")
chrom.all <- c("chr16")
dev.new()
sample <- "test"
for(chrom in chrom.all){
print(paste0("################## plot ", sample, " ", chrom, " #####################"))
# 保存为png
# png(filename = paste0(sample, "_", chrom, "_BAF_LRR.png"), width = 1200, height = 400)
# B.Allele.Freq Log.R.Ratio
# plot.type = 4, 在X轴方向绘制全部染色体
kp <- plotKaryotype(genome = genome.version,
plot.type = 4,
ideogram.plotter = kpAddCytobands,
labels.plotter = kpAddChromosomeNames,
plot.params = pp,
# cytobands = cytoband.GRanges,
chromosomes = chrom,
main=paste0("The B Allele Frequency and Log R ratio of ", chrom))
# kpAddBaseNumbers(kp)
kpAddBaseNumbers(kp, tick.dist=10000000, minor.tick.dist=2000000)
kpAddCytobandLabels(kp, cex = 0.8, force.all = TRUE)
# kpAddCytobandsAsLine(kp)
# kpAddCytobands(kp)
#################### BAF ####################
baf.r0 <- 0.4
baf.r1 <- 0.75
kpAxis(kp, r0 = baf.r0, r1 = baf.r1)
kpPoints(kp, data = snp.GRanges, cex=0.3,
y=snp.GRanges$BAF, r0=baf.r0, r1=baf.r1)
#################### LRR ####################
lrr.r0 <- 0.0
lrr.r1 <- 0.35
lrr.ymax <- 3
lrr.ymin <- -3
kpAxis(kp, tick.pos = c(lrr.ymin, 0, lrr.ymax),
r0=lrr.r0, r1=lrr.r1,
ymax=lrr.ymax, ymin=lrr.ymin)
kpPoints(kp, data = snp.GRanges, cex=0.3,
y=snp.GRanges$LRR, r0=lrr.r0, r1=lrr.r1,
ymax=lrr.ymax, ymin = lrr.ymin)
# 超过Y轴范围点显示为红色
if(length(lrr.below) > 0){
kpPoints(kp, data=snp.GRanges[lrr.below],
y=snp.GRanges[lrr.below]$LRR, cex=0.3,
r0=lrr.r0, r1=lrr.r1, ymax=lrr.ymax, ymin=lrr.ymin, col="red")
}
kpAbline(kp, h=0, r0=lrr.r0, r1=lrr.r1, ymax = lrr.ymax, ymin = lrr.ymin, col="red")
# c(0.95, 0.025, 0.025)
# kpPlotMarkers(kp, data = genes,
# labels = genes$hgnc_symbol,
# line.color = "blue",
# marker.parts = c(0.95), r1=1.05)
# dev.off()
}
3. 部分可视化图
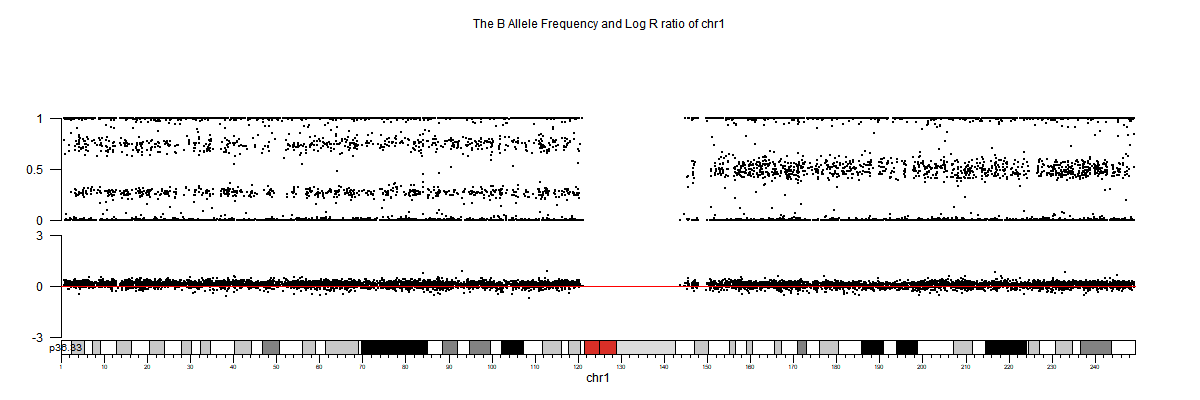
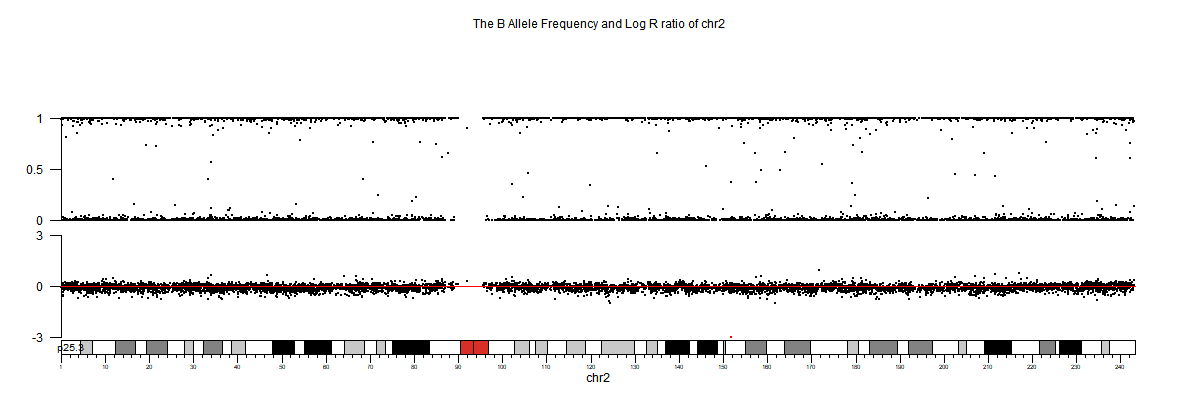
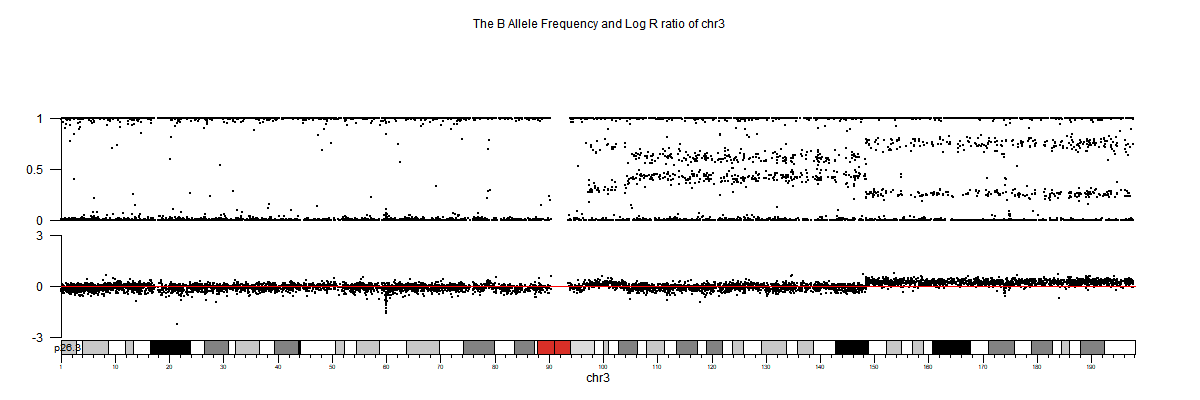

生信分析进阶文章推荐
生信分析进阶1 - HLA分析的HLA区域reads提取及bam转换fastq
生信分析进阶2 - 利用GC含量的Loess回归矫正reads数量
生信分析进阶3 - pysam操作bam文件统计unique reads和mapped reads高级技巧合辑


















 6386
6386

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











