









1.概念:
kmer(k)是一段固定长度的短序列,因为对于长度为L的序列(L>k),每次移动一个字符,共有N=L - k +1个kmer序列。此外,发现kmer在基因组大小评估和组装中为奇数,这是因为基因组的序列中存在回文序列。
2.用途:
用于基因组 De novo 前的基因组调查,评估基因组的size
公式:Genome_size = Total_kmer_number / Expected_coverage
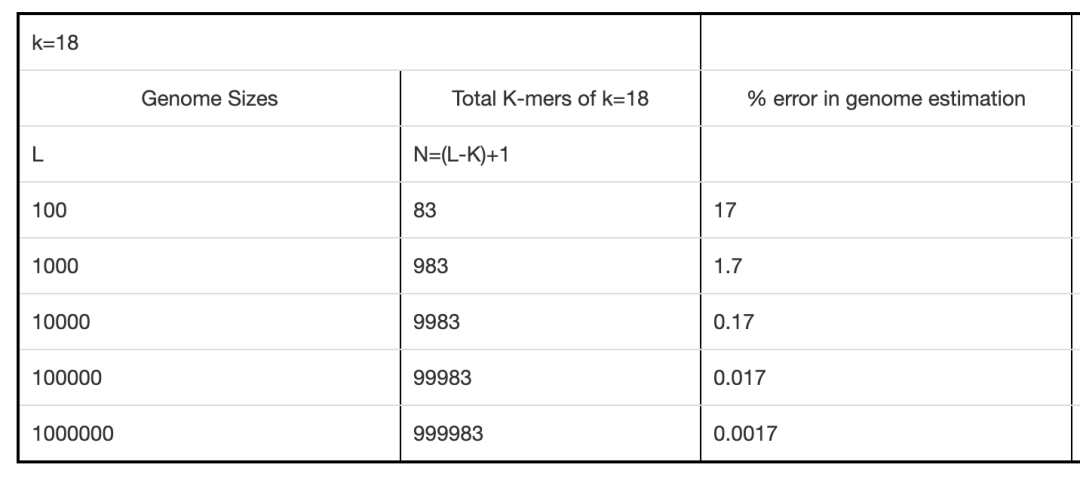
k-mer的总数与k值大小的关系
3.K-mer频数统计分布图
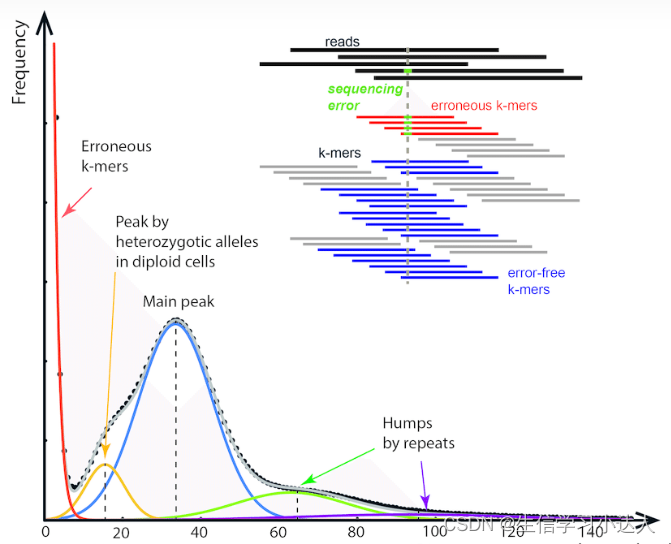
在测序覆盖均匀、不存在测序误差和基因组重复序列的理论条件下,K-mer分布曲线会符合泊松分布,存在一个主峰(Main peak主峰:单倍体或者纯合二倍体基因组)。如果基因组存在某些复杂特征,会使分布曲线偏离泊松分布,出现与特征相对应的峰(杂合体或多倍体)。
4. 评估软件Jellyfish
参看jellyfish官网(https://github.com/gmarcais/Jellyfish/blob/master/doc/Readme.md)
jellyfish count -t 8 -C -m 20 -s 10G -o K20 SRR8651760.1_1.paired.fastq.gz SRR8651760.1_2.paired.fastq.gz
参数:
-t -treads=unit32 Number of treads to be used in the run.
-C -both-strands Count both strands
-m -mer-len=unit32 Length of the k-mer
-s -size=unit32 Hash size / memory allocation
-o -output=string Output file name
##统计K-mer的histogram
jellyfish histo -t 10 jellyfish_kmer${kmer}.count.txt -o jellyfish_kmer$kmer.hist.txt
##GenomeScope软件可以将jellyfish结果拟合计算基因组杂合度信息
## 150 是PE read length
## ./genomescope_${kmer}_out 是输出文件夹
kmer=17
Rscript /home/debian/bin/genomescope-1.0.0/genomescope.R jellyfish_kmer${kmer}.hist.txt \
${kmer} 150 ./genomescope_${kmer}_out
This will count canonical (-C) 21-mers (-m 21), using a hash with 100 million elements (-s 100M) and 10 threads (-t 10) in the sequences in the file reads.fasta. The output is written in the file mer counts.jf by default (change with -o switch).
5.KmerGenie软件评估基因组大小
KmerGenie 1.7051
Usage:
kmergenie <read_file> [options]
Options:
--diploid use the diploid model (default: haploid model)
--one-pass skip the second pass to estimate k at 2 bp resolution (default: two passes)
-k <value> largest k-mer size to consider (default: 121)
-l <value> smallest k-mer size to consider (default: 15)
-s <value> interval between consecutive kmer sizes (default: 10)
-e <value> k-mer sampling value (default: auto-detected to use ~200 MB memory/thread)
-t <value> number of threads (default: number of cores minus one)
-o <prefix> prefix of the output files (default: histograms)
--debug developer output of R scripts
--orig-hist legacy histogram estimation method (slower, less accurate)6.利用 SOAPec
k-mer深度-频率分布使用SOAPec (v.2.0.1, https://sourceforge.net/projects/soapdenovo2/)生成,参数如下:-k 17 -q 33 -t 10。然后根据以下公式44计算基因组大小:基因组大小= k-mer覆盖率/平均k-mer深度。
如何选择合适的K-mer大小?一般我们会尝试不同长度K-mer的大小,或者使用一些工具帮助我们觉得K-mer大小,比如KmerGenie等软件。

















 5157
5157

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









