







前言
在之前的文章里写了安装infercnv包的方法,后续运行该包的过程中,遇到了一些问题,现将问题及解决办法记录下来,以供大家参考,如有不足,烦请各位指正,谢谢!
问题一:关于输入文件
在运行infercnv之前,需要准备三个文件:1.单细胞表达矩阵文件;2.细胞类型注释文件;3.基因坐标文件。三个文件的样子
1.表达矩阵文件
行名为基因名,列名为细胞样本名,表达矩阵必须为数值型矩阵:
示例的表达矩阵:

查看一下示例文件的矩阵类型:
raw_counts_matrix="/Documents/R/win-library/4.0/infercnv/extdata/oligodendroglioma_expression_downsampled.counts.matrix.gz"
if (substr(raw_counts_matrix, nchar(raw_counts_matrix)-2, nchar(raw_counts_matrix)) == ".gz") {
raw.data <- read.table(connection <- gzfile(raw_counts_matrix, 'rt'), sep="\t", header=TRUE, row.names=1, check.names=FALSE)
close(connection)
raw.data <- as.matrix(raw.data)}
str(raw.data)
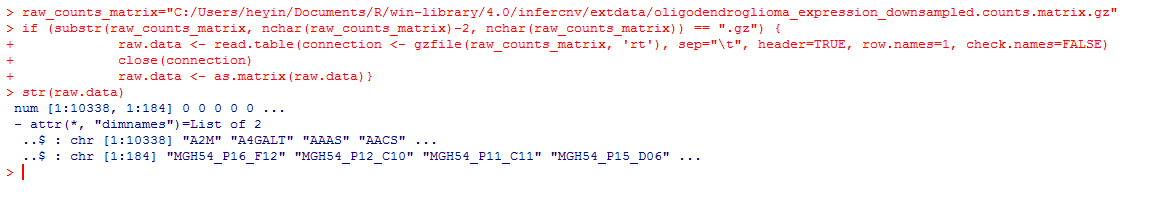
如果表达矩阵不是数值型,会在后续运行报错:

因为矩阵只能有一种数据型,故在infercnv自行转换为矩阵时,把表达矩阵转换成了字符型矩阵,这里提出一种解决办法:
#创建没有行名列名的表达矩阵
dat=read.table("/mix_exp_M.csv",stringsAsFactors=FALSE, header=TRUE 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章
















 1万+
1万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









