







InferCNV可以用于肿瘤单细胞RNA-Seq数据中鉴定大规模染色体拷贝数变异,例如整个染色体或大片段染色体的扩增或缺失。基本思路是在整个基因组范围内,将每个肿瘤细胞基因表达与平均表达或“正常”参考细胞基因表达对比,确定其表达强度。
1. 安装
BiocManager::install("infercnv")
library("infercnv")如果遇到安装问题可以参考infercnv安装遇到的问题。
2. 输入
在进行分析之前,需要准备三个文件:
- 单细胞表达矩阵文件
- 细胞类型文件
- 基因在染色体上的位置信息文件
基因在染色体上的位置信息文件第一列为gene symbol名称,第二列是染色体号,后两列是基因的始末位置,制表符分隔。格式如下:

这个文件很多只是说了里面的具体内容但是没有说怎么得到的,我也是花了一段时间才知道怎么得到的。
1.一些基因组位置文件是从共同参考文献生成的,并在TrinityCTAT上提供。下载你需要的文件。
2.构建自己自定义的:
1)CMD端获取最新的gtf文件:
wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_35/gencode.v35.annotation.gtf.gz2)Python运行:先从infercnv的github上https://github.com/broadinstitute/infercnv/blob/master/scripts/gtf_to_position_file.py下载gtf_to_position_file.py脚本,存为.py后缀的文件,运行
python gtf_to_position.py --attribute_name "gene_name" gencode.v35.annotation.gtf gene_pos.txt输出文件gene_pos.txt就是我们想要的基因位点数据文件。
因为我的数据已经用Seurat处理过了,所以我直接用Seurat对象读取另外两个文件
library(infercnv)
pbmc <- readRDS('../data/pbmc.RDS')
table(pbmc$seurat_clusters)
celltype <- cbind(colnames(pbmc),as.character(pbmc@meta.data$seurat_clusters))
head(celltype)
write.table(celltype,file = "celltype_pbmc.txt",
sep = '\t',row.names=F,col.names=F,quote=F) # 准备细胞类型文件
mtx <- pbmc@assays$RNA@counts
3. 使用
infercnv_obj = CreateInfercnvObject(
raw_counts_matrix = mtx,
annotations_file = 'celltype_pbmc.txt',
gene_order_file="gencode_v19_gene_pos.txt",
ref_group_names=c("4",'8'))
infercnv_obj = infercnv::run(infercnv_obj,
cutoff=0.1,
# use 1 for smart-seq, 0.1 for 10x-genomics
out_dir = 'infercnv/',# dir is auto-created for storing outputs
cluster_by_groups=T,
# 选择TRUE是按样本分组 改为FALSE会进行按另一个参数k_obs_groups给出的分组数(默认为1)进行分组
denoise=F,
HMM=F, # 是否基于HMM预测CNV,True的话时间很久
no_prelim_plot=TRUE,
analysis_mode ='subclusters' # 区分肿瘤亚型,慢
)4 输出
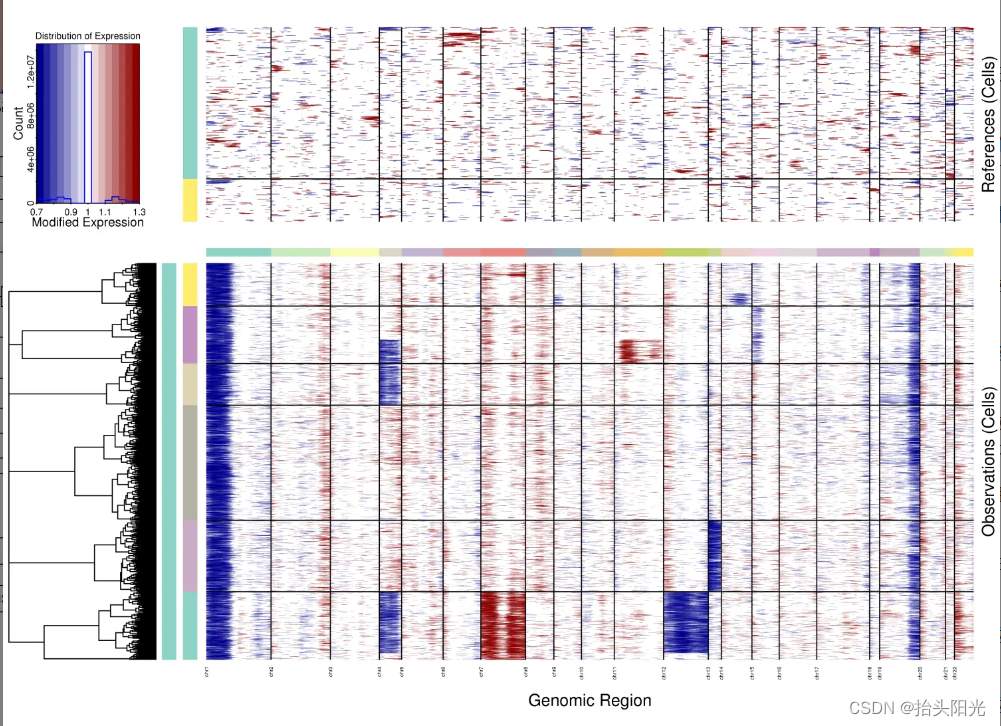
我认为重要的输出文件infercnv.png
降噪之后生成的最终热图,图中的数值是"Residual expression values",可以简单理解为基因表达值的另一种形式;
5. 画图
infercnv包也包含了画图函数plot_cnv,这个使用的人不多
library(RColorBrewer)
infercnv::plot_cnv(infercnv_obj, #上两步得到的infercnv对象
plot_chr_scale = T, #画染色体全长,默认只画出(分析用到的)基因
output_filename = "better_plot",output_format = "pdf", #保存为pdf文件
custom_color_pal = color.palette(c("#8DD3C7","white","#BC80BD"), c(2, 2))) #改颜色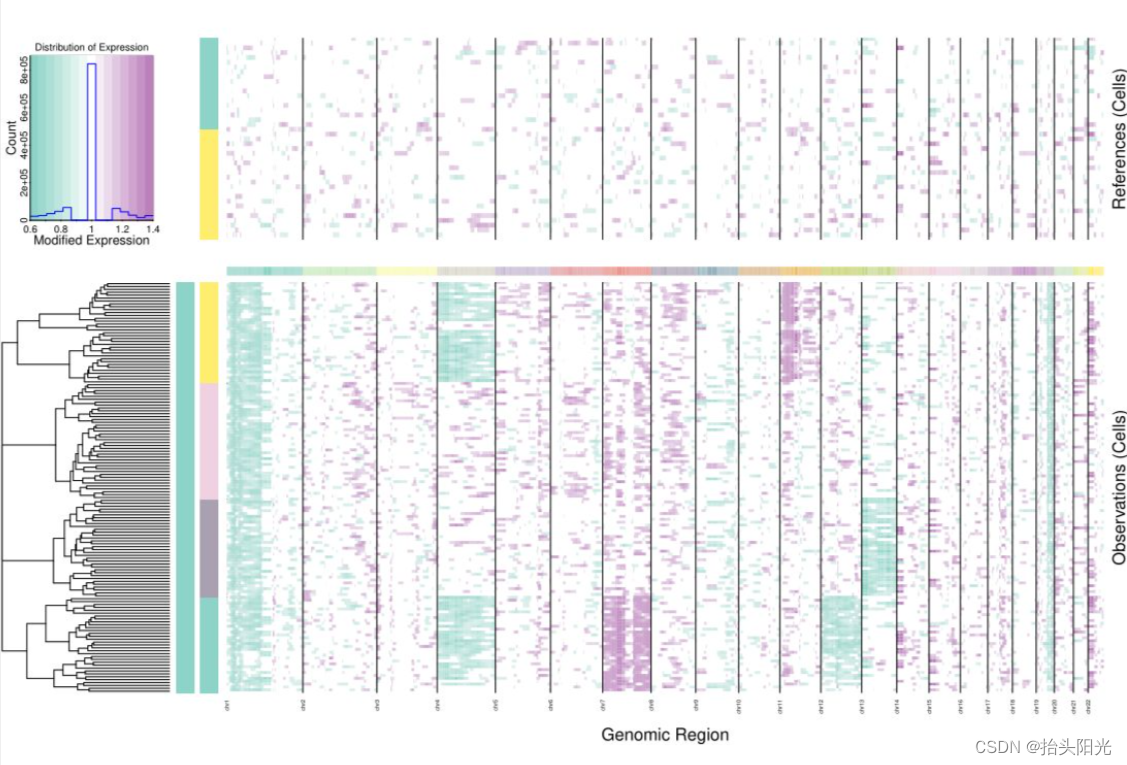















 1203
1203











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









