







几种Featurizer解读
Deepchem的Featurizer我倾向于翻译成‘特征提取器’。我们都知道化学分子本身不能直接作为机器学习的读入数据,这就需要我们将其结构转变为数字形式。而featurizer可以很方便做到这一点,它可以将化学分子(SMILE格式)转变为长度一致的向量或维度一致的矩阵形式。
为了方便说明,我们需要提前准备好需要进行特征提取的小分子:
import deepchem as dc
smiles = [
'O=Cc1ccc(O)c(OC)c1',
'CN1CCC[C@H]1c2cccnc2',
'C1CCCCC1',
'c1ccccc1',
'CC(=O)O',
]
CircularFingerprint
从字面意义看,这种特征提取方式或多或少与‘圆’有关。事实正是如此,如下图所示,对于化学分子中任意一个原子,以其为中心绘制不同半径的同心圆,根据这些同心圆包含的子结构赋值不同的数字,这些数字按顺序排列成相同长度的向量。生成的向量可以作为机器学习的读入数据。
:::
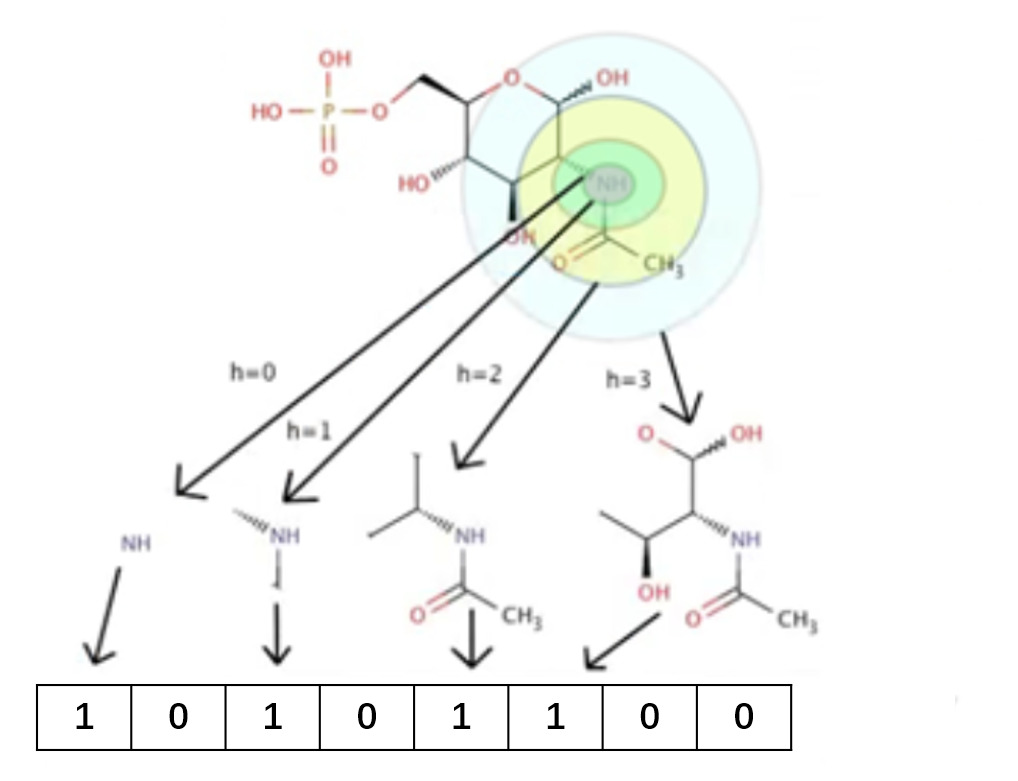
对应代码如下所示:
#初始化特征提取器
featurizer = dc.feat.CircularFingerprint()
#读入需要提取特征的分子
feature = featurizer.featurize(smiles)
#这种方法生成的每个分子向量长度默认均为1024。
RDKitDescriptors
这种类型的特征提取器调用底层的rdkit模块计算分子描述符(描述符可以包括分子量、溶剂可及表面积、氢键供/受体数量等)。
#初始化特征提取器
rdkit_featurizer = dc.feat.RDKitDescriptors()
#读入需要提取特征的分子
rdkit_feature = rdkit_featurizer.featurize(smiles)
#这种方法生成的每个分子向量长度默认均为208。
WeaveFeaturizer and MolGraphConvFeaturizer
这两种方法比较特殊,都是将分子转化为二维图形进而提取特征值。如果你事先了解过图形的卷积神经网络或许好理解一些。
以MolGraphConvFeaturizer调用为例:
#初始化特征提取器
graph_featurizer = dc.feat.MolGraphConvFeaturizer()
#读入需要提取特征的分子
graph_feature = graph_featurizer.featurize(smiles) 。
CoulombMatrix
- 以上介绍的分子特征生成器都是基于分子内在自有属性生成的;我们知道小分子在结合受体分子时往往采用各种构象,如果想要准确预测它们之间的结合强度那就不得不考虑分子可能产生的各种构象。
- 直白翻译CoulombMatrix可以知道这种分子特征生成器和库伦相互作用有关。两个电荷之间的静电库仑相互作用与电荷的位置和距离成正比(满足q1q2/r)。q1、q2表示两个原子所带电荷,r表示两原子间距离。
- 所谓库伦矩阵,是对于一个分子的所有原子(总数N),生成N*N的矩阵,矩阵中的每个元素给出了任意两原子间的库仑相互作用。
调用方法:
from rdkit import Chem #导入rdkit模块用于生成分子构象
#初始化分子构象生成器(定义最大构象生成个数)
generator = dc.utils.ConformerGenerator(max_conformers=5)
#读入目标分子的smiles式
for m in smiles:
mol = generator.generate_conformers(Chem.MolFromSmiles(m))
print(len(mol.GetConformers()))
#生成库伦矩阵
coulomb_mat = dc.feat.CoulombMatrix(max_atoms=20)
#max_atoms参数项是为了让所有分子获得同样大小的矩阵,这样利于输入训练模型
features = coulomb_mat(mol)
最后每个分子的每个构象生成的features都是一个20*20的矩阵
CoulumbMatrixEig
CoulombMatrixEig继承自CoulombMatrix, 它首先计算不同分子的不同构象的库仑矩阵,然后计算每个库仑矩阵的特征值或者说特征向量。最后填充这些特征值,以减轻分子中原子数量的变化对训练结果的影响。
调用方法:
#初始化CoulumbMatrixEig
coulomb_mat_eig = dc.feat.CoulombMatrixEig(max_atoms=20)
#读入分子
features = coulomb_mat_eig(smiles)
扫描下方二维码,关注公众号获得更多实用干货:

















 3670
3670











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











