






 Foldseek是一个新的结构搜索程序,能快速找到三维结构相似的蛋白质,帮助研究人员鉴定未知蛋白质的功能。通过将蛋白质结构转换为“结构字母表”,Foldseek结合了基于结构和序列搜索的优点,提高了搜索效率和准确性。这款开源软件已经在多个结构数据库中得到应用,对于生物学研究,尤其是新病原体蛋白质功能的确定具有重大意义。
Foldseek是一个新的结构搜索程序,能快速找到三维结构相似的蛋白质,帮助研究人员鉴定未知蛋白质的功能。通过将蛋白质结构转换为“结构字母表”,Foldseek结合了基于结构和序列搜索的优点,提高了搜索效率和准确性。这款开源软件已经在多个结构数据库中得到应用,对于生物学研究,尤其是新病原体蛋白质功能的确定具有重大意义。
Foldseek为结构搜索程序,它可以轻松找到三维结构相似的蛋白质。
Foldseek允许研究人员鉴定结构类似的蛋白质。图片来源:DeepMind
当你发现一种蛋白质时,如何确定其功能?这正是Gregory Gloor面临的问题。
Gregory Gloor是加拿大伦敦西安大略大学(University of Western Ontario in London, Canada)的生物化学家,Gloor正在研究油田废水处理厂的细菌群落,希望能鉴定有助于细菌分解有毒物质的蛋白质。他开始研究感染这些细菌的病毒(称为噬菌体)表达的蛋白质。遗憾的是,在已知的蛋白质数据库中未查询到任何结果。
然后,Gloor找到一款名为Foldseek的搜索工具,其创建者于2021年首次共享,并于2023年5月8日在线发表于《Nature Biotechnology》杂志。他说:“这就像‘哈利路亚’。”他的项目“从基本不可能成为了可能”。
蛋白质由氨基酸组成的多肽链折叠而成,其折叠结构决定其功能。在过去几年里,仅从氨基酸序列推断蛋白质三维结构的人工智能工具有了巨大提高。研究人员使用Google DeepMind开发的AlphaFold 2、华盛顿大学西雅图校区团队的RoseTTAFold和其他此类工具编译并创建了包含数亿个蛋白结构的数据库。Foldseek可以快速搜索这些数据库,找到与目标蛋白质结构相似的蛋白质——显然,功能也相似。
两全其美
寻找氨基酸序列相似的蛋白质是确定不熟悉蛋白质功能的常规算法。如果这些相关蛋白质的功能已知,研究人员可以预测新蛋白质的功能。
基于序列的搜索,速度很快,就像在硬盘上搜索文件名。但它们通常会错过很好的匹配,因为结构相似的蛋白质可以有完全不同的序列。基于结构的搜索,寻找结构相似而不是序列相似,但这可能需要数千倍的时间,因为比较复杂的三维结构从算法上来讲是很困难的。借助Foldseek,研究人员得到了两者的最佳结合:该软件将蛋白质的结构表示为一串字母,即“结构字母表”,从而既提供了基于结构搜索的灵敏度, 又具备了基于序列的搜索的速度。
首尔国立大学的生物学家Martin Steinegger是该论文通讯作者。他说:“关键思想之一:为了产生良好的结构搜索结果,正确编码非常重要。”
ColabFold通过将MMseqs2的快速同源搜索与AlphaFold2或RoseTTAFold相结合,提供了对蛋白质结构和复合物的加速预测。Gloor首先使用ColabFold预测他发现的噬菌体蛋白质的结构,然后使用Foldseek与已知蛋白质数据库进行匹配。结果发现,其中一些蛋白质是形成病毒的外壳;其他的是酶。他的评价:Foldseek“非常聪明”。
Foldseek并非第一个将蛋白质结构简化为字母表的算法。其他搜索工具通常根据蛋白质序列中紧接其前和后氨基酸的方向,给每个氨基酸分配一个字母。然而,这种方法忽略了线性链中相距较远但在三维空间中相邻的氨基酸之间的相互作用。Foldseek根据氨基酸与其在折叠的蛋白质中最接近的氨基酸的距离和定位来为每个氨基酸分配20个字母中的一个。Steinegger表示,通过关注这些空间桥梁,Foldseek的“三维相互作用字母表”更好地捕捉了蛋白质的全局结构。
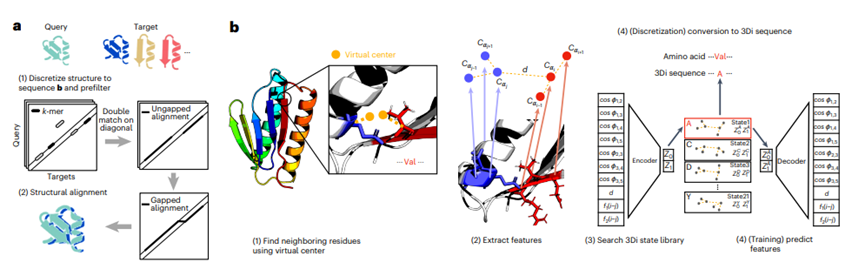
Foldseek工作流程:a. Foldseek在一组查询结构和一组目标结构中进行搜索。(1)将查询和目标结构离散化为3Di序列。为了检测候选结构,我们对3Di序列应用了MMseqs2的快速和敏感的k-mer和无间隔对齐预过滤器。(2)通过结合3Di和氨基酸替代得分的向量化Smith-Waterman局部对齐来进行筛选。或者,使用加速了1.7倍的TM-align版本进行全局对齐。b. 学习3Di字母表。(1)3Di状态描述了残基i与其最近邻j之间的三维相互作用。最近邻具有最接近的虚拟中心距离(黄色)。虚拟中心位置经过优化以获得最大的搜索灵敏度。(2)我们从两个主链片段(蓝色和红色)的六个Cα坐标中提取七个角度、欧几里德Cα距离和两个序列距离特征,以描述残基i和j之间的相互作用几何形状。(3)使用VQ-VAE28进行训练来定义20个3Di状态,这些状态在进化上是最大程度保守的。在结构搜索中,编码器预测每个残基的最佳匹配3Di状态。
回顾过去
欧洲分子生物学实验室欧洲生物信息学研究所(European Molecular Biology Laboratory’s European Bioinformatics Institute)的计算生物学家Janet Thornton说:“生物学发生在三维空间中。” 基于蛋白质结构的比较"使您能够更远地观察进化时间,从而可以识别出从同一前体蛋白质进化而来的非常遥远的亲属"。
为了测试Foldseek,Steinegger的团队使用了一个包含365,000万个蛋白质的数据库,这些蛋白质的结构是由AlphaFold 2预测所得。他们将其中的100个结构输入Foldseek,并要求它为每一个结构在数据库中查询排名最相似的蛋白质。分数是根据算法检索到的"真阳性"的数量(即根据原子模型得分高于某个相似性阈值的蛋白质)以及检索到"假阳性"之前的数量来确定的。与两种常见的基于结构的搜索工具TM-align和Dali相比,Foldseek的表现更优,分别提高了24%和8%,速度快提高了35000和20000倍。与基于结构字母表的工具CLE-SW相比,Foldseek提高了23%,速度提高11倍[1]。
Foldseek作为开源软件适用于macOS和Linux计算机。开发者还创建了一个Web服务器, 供研究人员搜索涵盖数亿个蛋白质的七个结构数据库中的任意一个。据Steinegger称,该软件至少被安装了14000次,研究人员每天在服务器上运行约300次搜索。
Thornton表示,Foldseek可以帮助研究人员确定新病原体中蛋白质的功能,或者简单地揭示生物体的运作方式。例如,Steinegger和他的团队应用Foldseek在AlphaFold数据库中找到相关蛋白质簇,并确定了一种与人类组蛋白结构相似的细菌蛋白质[3]。
利用现有的搜索工具,Gloor找到了他研究中一小部分噬菌体蛋白质的匹配项,但这些噬菌体蛋白都没有已知的功能。使用Foldseek工具,他找到了一半的蛋白质匹配项,并将其中15%确定为酶[2]。
Gloor表示:“将三维相互作用的结构转化为字符串需要相当程度的洞察力和独创性。”使用Foldseek,科学家可以了解更多生物体中的更多蛋白质。“这将改变做进化研究的方式和方法,”他说。“它将提高我们寻找真正独特的生态系统并找出其工作原理的能力。”
参考文献:
[1]. van Kempen, M. et al. Nature Biotechnol. https://doi.org/10.1038/s41587-023-01773-0 (2023).
[2]. Say, H., Joris, B., Giguere, D. & Gloor, G. B. Preprint at bioRxiv https://doi.org/10.1101/2023.04.19.537516 (2023).
[3]. Hernandez, I. B. et al. Preprint at bioRxiv https://doi.org/10.1101/2023.03.09.531927 (2023).
阅读原文内容:
https://www.nature.com/articles/d41586-023-02205-4
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

















































 2663
2663

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











