






 文章介绍了如何利用pymol软件生成含有蛋白二级结构信息的txt文件,然后通过处理这个文件得到二级结构的位置,如alpha-helix和beta-sheet。接着,文章提供了一个方法整理这些数据成特定格式,并推荐了一个在线网站(novopro.cn)来创建蛋白质序列和二级结构的图形表示。此外,还提到了其他在线工具可用于比较不同蛋白质的二级结构。
文章介绍了如何利用pymol软件生成含有蛋白二级结构信息的txt文件,然后通过处理这个文件得到二级结构的位置,如alpha-helix和beta-sheet。接着,文章提供了一个方法整理这些数据成特定格式,并推荐了一个在线网站(novopro.cn)来创建蛋白质序列和二级结构的图形表示。此外,还提到了其他在线工具可用于比较不同蛋白质的二级结构。
很多时候我们想要展示蛋白质二级结构的多少和位置,如下简单介绍了两种可以实现此要求的方法。
一、在pymol中输出含有蛋白二级结构信息的文件
load AB.pdb
select all
open("potein.txt","w").writelines( ["Residue %s: %s\n"%(a.resi,a.ss) for a in cmd.get_model("AB" +" and n. ca").atom] )
delete all打开生成txt文本,每行(每个氨基酸)最后一个字符表示不同二级结构的简写,其中H表示alpha-helix,L表示loop (coil),S表示 beta-sheet。
二、获取所有的二级结构并整理成如{'L': '1-5,10-11,16-18', 'S': '6-9,12-15'}样式
f = open("protein.txt","r")
lst = []
for a in f.readlines():
lst.append(a[-3:-1])
def format_list(lst):
output = {}
start = None
prev_char = None
for i, char in enumerate(lst):
if char != prev_char:
if prev_char is not None:
end = i - 1
if start < end:
output[prev_char] = output.get(prev_char, []) + [(start, end)]
else:
output[prev_char] = output.get(prev_char, []) + [start]
start = i
prev_char = char
# handle the last character sequence
if start is not None:
end = len(lst) - 1
if start < end:
output[prev_char] = output.get(prev_char, []) + [(start, end)]
else:
output[prev_char] = output.get(prev_char, []) + [start]
# format the output as a string
result = {}
for k, v in output.items():
s = ""
for seq in v:
if isinstance(seq, tuple):
s += f"{seq[0]+1}-{seq[1]+1},"
else:
s += f"{seq+1},"
result[k] = s.rstrip(",")
return result三、将上述字典结果复制到在线网站作蛋白质序列和二级结构图
打开在线网站:https://www.novopro.cn/tools/fancy-ss.html
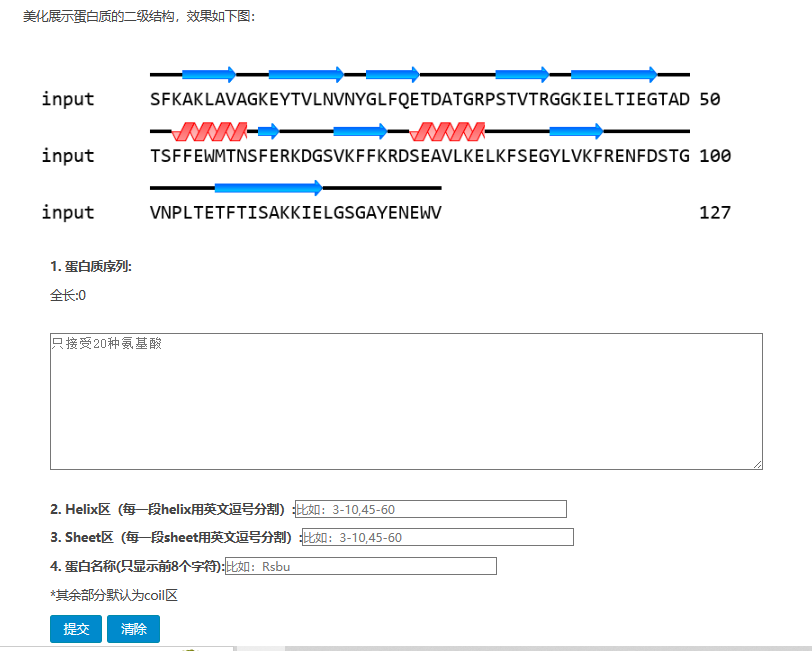
四、其他在线网站
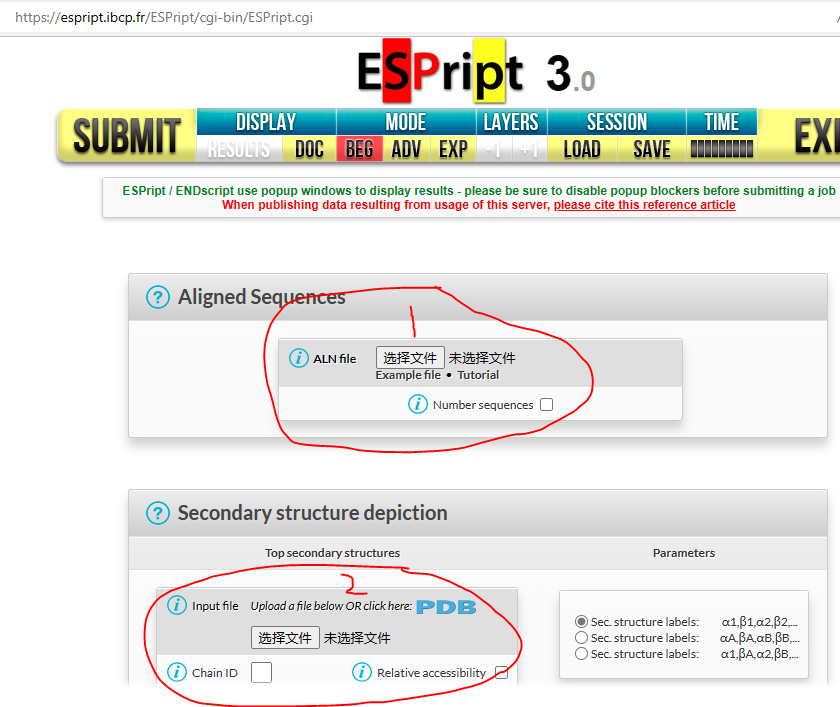
在上图1中添加含有蛋白质序列比对结果格式的文件,在图2添加任意一个蛋白质的PDB文件,可以两次分别输入不同的蛋白以获得两个蛋白的二级结构。

















 626
626

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









