







tradeSeq是一个可以沿着轨迹分析基因表达的R包,也可以用来分析特定pathway沿轨迹的表达,具体方法见后续分享。
1.准备文件
library(BiocParallel)
library(tradeSeq)
#load slingshot sds文件
load(file = "./scsds.RData")
#load sc counts
load(file = "./sccounts.RData")
pseudotime <- slingPseudotime(sds, na = FALSE)
cellWeights <- slingCurveWeights(sds)
crv <- SlingshotDataSet(sds)
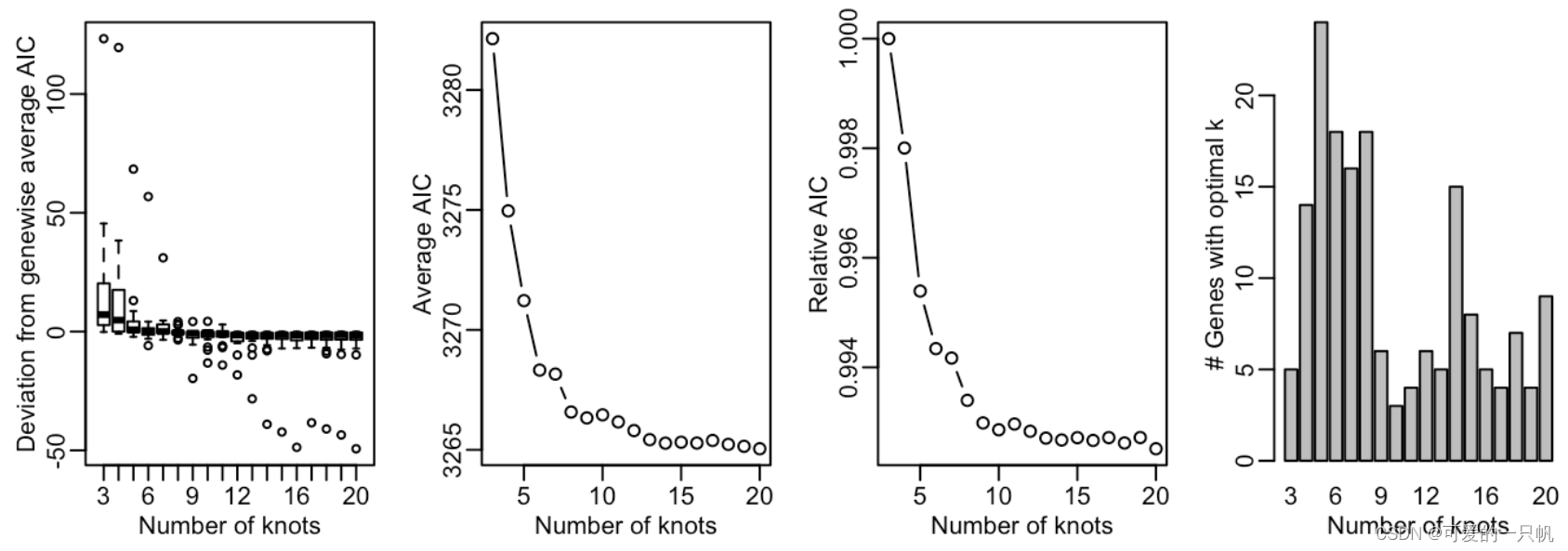
2.拟合负二项式模型确定nknots
icMat <- evaluateK(counts = counts,
sds = crv, nGenes = 200,
k = 3:10,verbose = T, plot = TRUE)
sce <- fitGAM(counts = counts,
pseudotime = pseudotime,
cellWeights = cellWeights,
nknots = 6, verbose = TRUE,
BPPARAM = MulticoreParam(20),parallel=T)
table(rowData(sce)$tradeSeq$converged)#查看收敛基因个数
3.谱系内比较
startRes <- startVsEndTest(sce,lineages=T)
oStart <- order(startRes$waldStat, decreasing = TRUE)
sigGeneStart <- names(sce)[oStart[1]]
plotSmoothers(sce, counts, gene = sigGeneStart)
plotGeneCount(crv, counts, gene = sigGeneStart)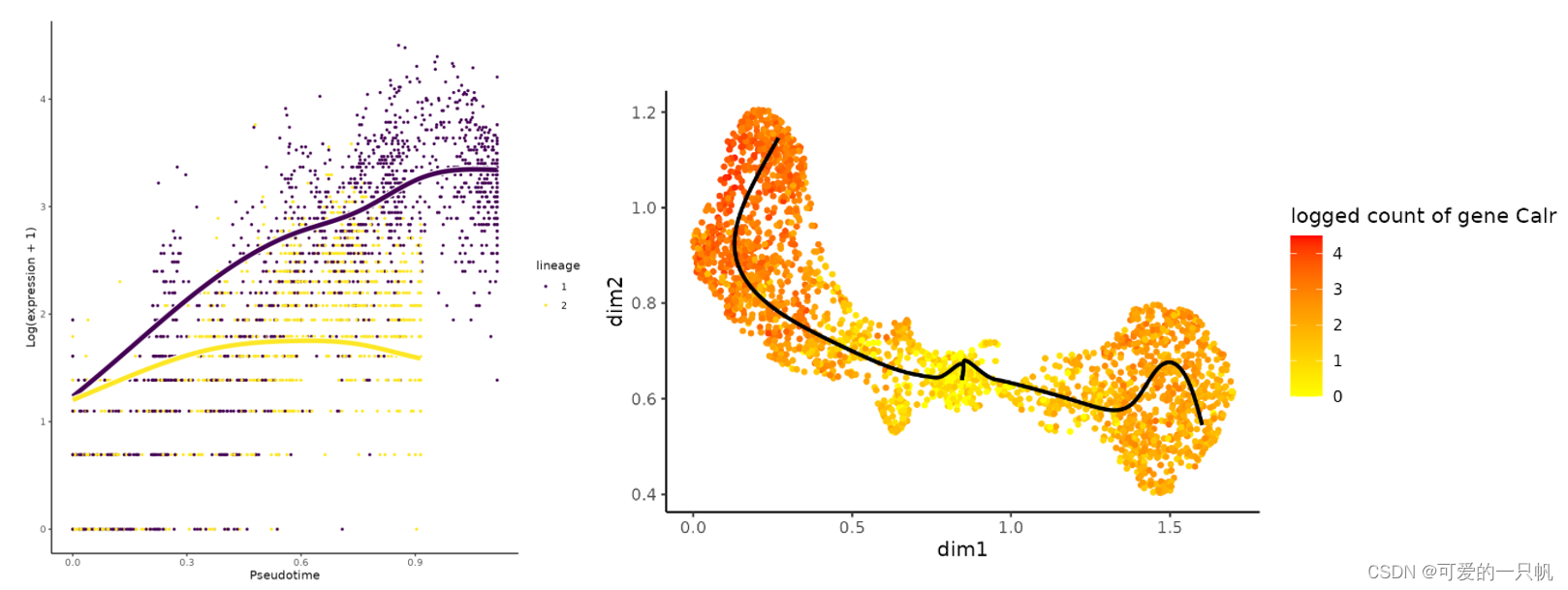 4.谱系间比较
4.谱系间比较
endRes <- diffEndTest(sce,pairwise=T)
o <- order(endRes$waldStat, decreasing = TRUE)
sigGene <- names(sce)[o[1]]
plotSmoothers(sce, counts, sigGene)
plotGeneCount(crv1, counts, gene = sigGene)出的图和上面类似,接下来可以筛选谱系间和谱系内都显著表达的基因,确定每个谱系特异表达的基因。
#Lineage1
tmp1 <- startRes[startRes$pvalue_lineage1<0.01,] %>% rownames()
tmp2 <- endRes[endRes$pvalue_1vs2<0.01,]%>%rownames()
intersect(tmp1,tmp2)

















 2535
2535

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









