






第一步:准备基因组文件
假如需要划分的窗口的参考基因组为hg19,可参考bedtools说明中的方法远程连接UCSC的数据库,提取相应的染色体和长度

得到的genome.txt:两列分别为染色体名称和对应长度,用tab间隔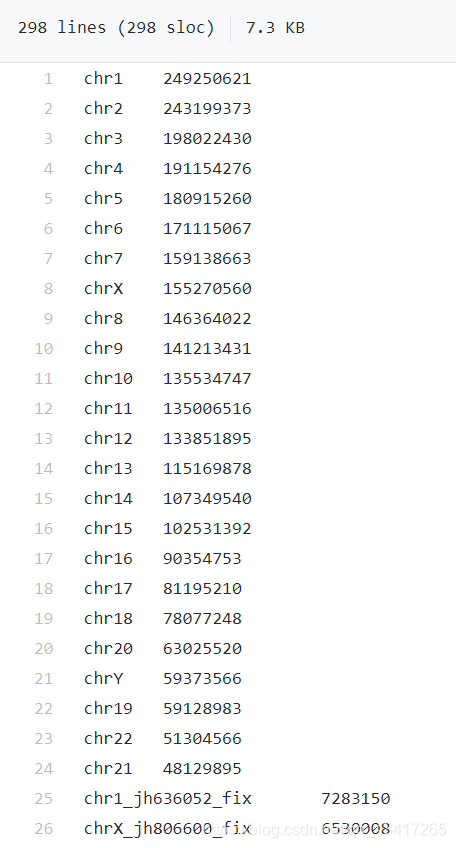
第二步:划分窗口
bedtools makewindows -g genome.txt -w 1000 > windows.bed
- -g genome.txt是要划分的基因组,格式为两列:染色体、染色体长度
- -w 1000为窗口大小为1k
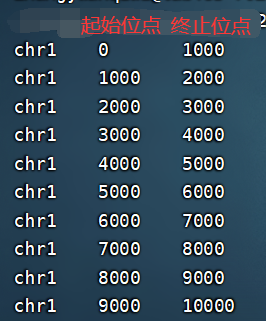
- windows.bed为输出文件,格式为三列:染色体、区间开始位点、区间结束位点。
- 生成的bed文件:
第三步:统计窗口内的平均覆盖深度
bedtools coverage -a windows.bed -b SRR081241.sorted.bam > HG00096.depth.txt
bedtools coverage对划分好的每个滑动窗口进行reads数(depth)的统计。
- -a windows为上一步划分好的区间
- -SRR081241.sorted.bam为测序数据mapping到参考基因组的比对文件
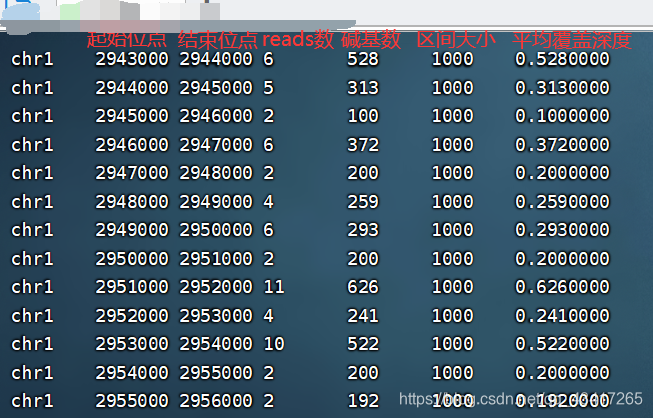
- HG00096.depth.txt为统计结果的输出文件,格式为7列:染色体、区间起始位点、区间结束位点、该区间内的reads数、该区间内的碱基数、区间大小、该区间的平均覆盖度
- 生成的txt文件:

















 2702
2702

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









