







叶绿体基因组组装
getorganelle组装叶绿体基因组
手把手注释教学
B站视频教程
安装
conda install -c bioconda getorganelle
安装叶绿体基因组databases
get_organelle_config.py --add embplant_pt
NCBI测试数据下载
从网上随机找了一篇文章 进行组装
文章链接
# download data
prefetch SRR15255748.sra
# split files
fasterq-dump --split-3 SRR15255748.sra
# after split
# SRR15255748_1.fastq SRR15255748_2.fastq
软件运行
get_organelle_from_reads.py -1 SRR15255748_1.fastq -2 SRR15255748_2.fastq -k 21,77,127 -o results -t 30 -R 25 -F embplant_pt
组装结果
使用Bandage 对 gfa/fastg 结果文件进行查看
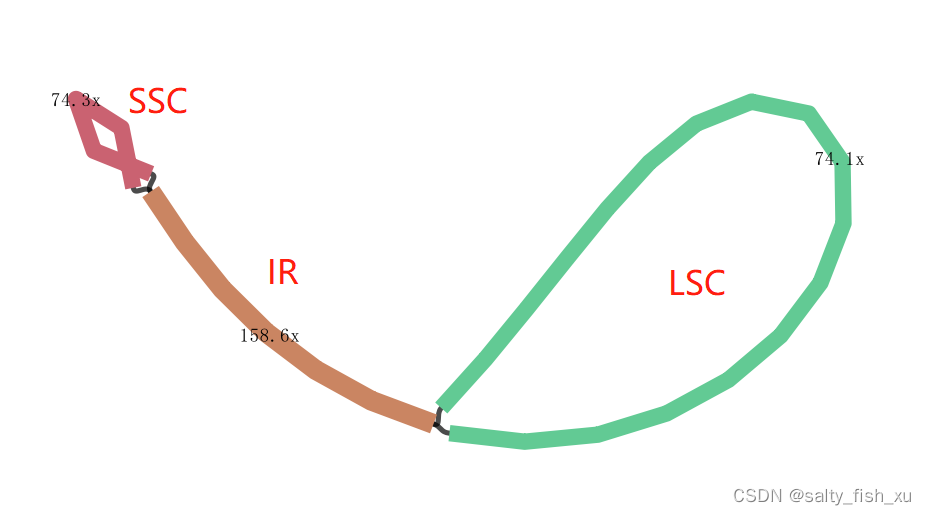
IR区域 拆分之后 就可以行成叶绿体的环状结构
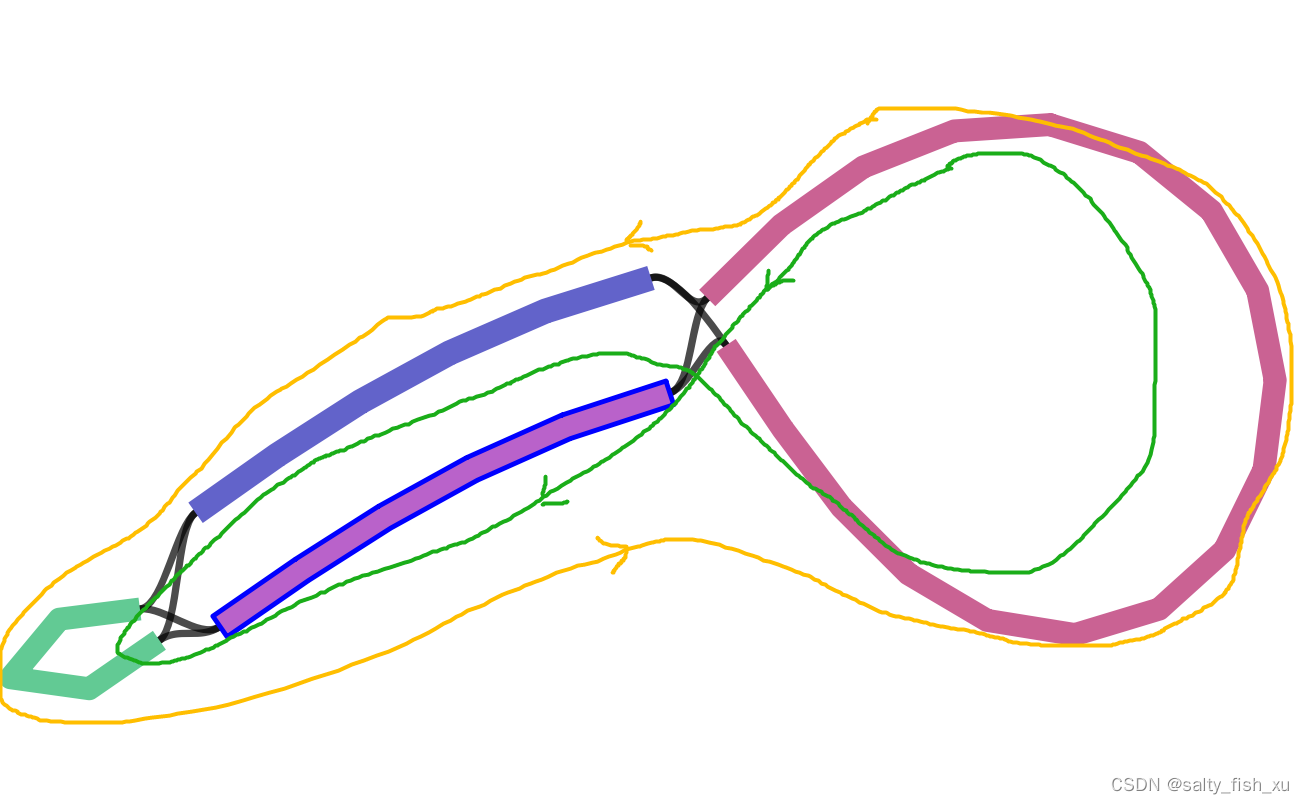
IR拆分之后就行成了两种成环方式,这也是为什么getorganelle会给出两条序列的原因,两条序列的差异在于SSC的方向。 因此我们需要对得到的两条序列做共线性分析。选择一条和参考相同的,或者NCBI用的比较多的一种方式 进行下一步分析。
选用参考自己做组装
参考序列下载
选用近缘的物种作为参考物种 这里选用 NC_063470.1 做为参考物种
数据处理
# build index
bowtie2-build test.fa ref # test.fa NC_036134.1
# mapping
bowtie2 -x ref --very-sensitive-local -1 SRR15255748_1.fastq -2 SRR15255748_2.fastq > mapping.sam
# sam to bam
samtools view -h -F 4 -@ 6 mapping.sam > mapping.bam
# bam to fastq
samtools fastq -1 1.fq -2 2.fq -s unmapped.fq mapping.bam
SPAdes组装
spades.py -k 21,77,127 -1 1.fq -2 2.fq -t 30 -o results
组装结果
使用Bandage 对 gfa/fastg 结果文件进行查看
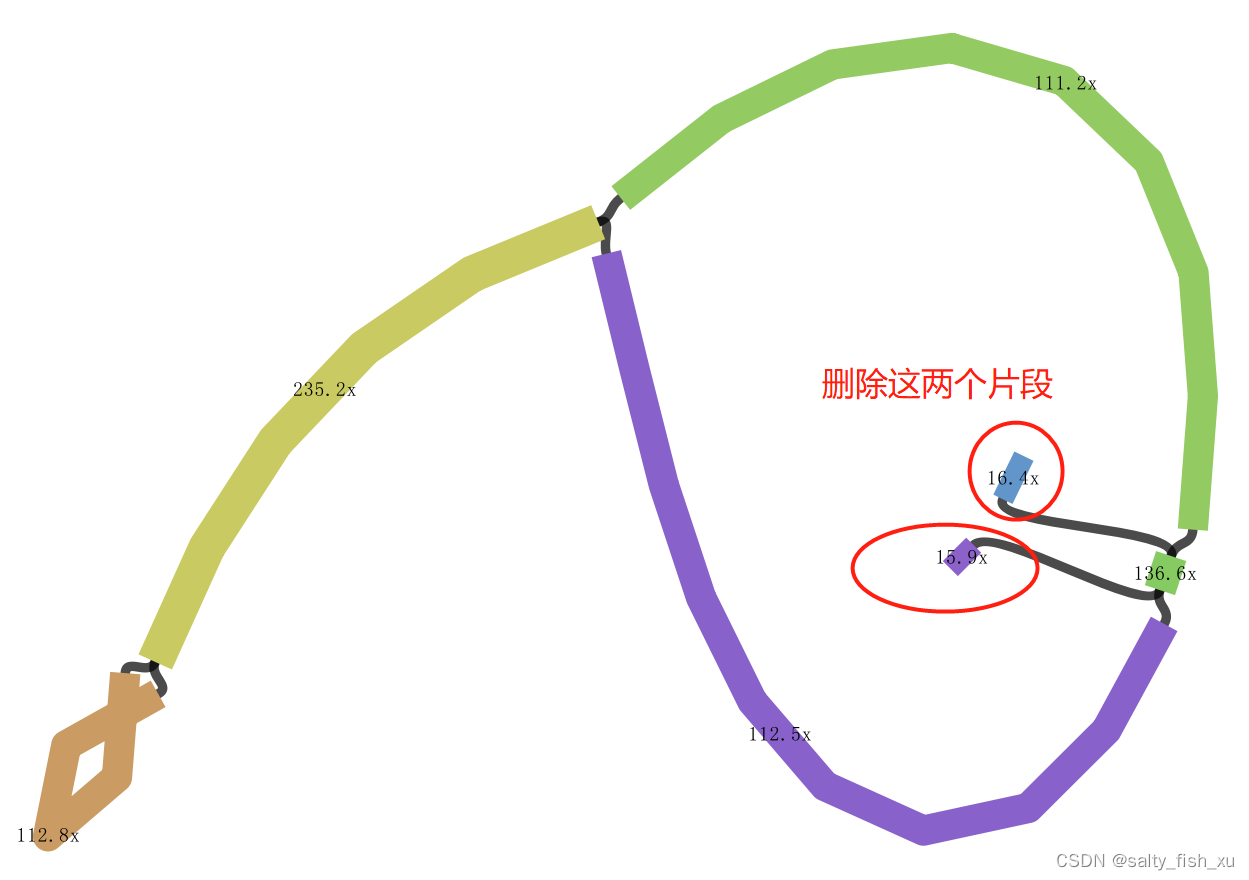
然后按照上面的处理方式 就可以得到相同的结果 如果序列前端和序列后端有一条Kmer是相似的 需要删除该Kmer
写在结尾
序列组装好之后 鉴定叶绿体的四个区域,然后序列调整LSC的第一个碱基 作为序列的开始, 接着就可以进行注释等后续分析。
当序列复杂度比较高时,使用getorganelle也是无法成环或者成环数比较多时,需要自己进行纠正!!!
如果有疑问,或者组装有问题 可以私信我,我看到一般都会回复的, 如果有帮助 记得帮忙点赞哦
参考链接:
https://github.com/Kinggerm/GetOrganelle
https://www.bilibili.com/video/BV1vq4y1g7QB/?spm_id_from=333.999.0.0

















 969
969

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









