









# sam or bam input
$ samtools stats test.sam > test_sam_stat.txt
下图为示例统计的SN关键字部分:
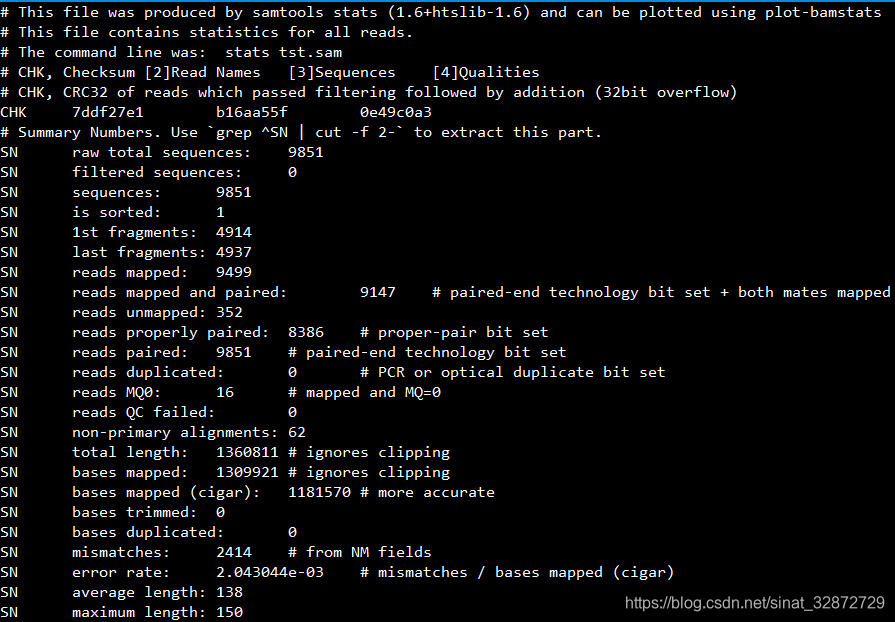
统计结果中包含的信息有:
| 关键字 | 官方解释 | 说明 |
|---|---|---|
| CHK | Checksum | 校验和 |
| SN | Summary numbers | 摘要编号 |
| FFQ | First fragment qualities | Read1片段质量 |
| LFQ | Last fragment qualities | Read2片段质量 |
| GCF | GC content of first fragments | Read1的GC含量 |
| GCL | GC content of last fragments | Read2的GC含量 |
| GCC | ACGT content per cycle | 每个cycle的ACGT含量 |
| FBC | ACGT content per cycle for first fragments only | Read1片段每个cycle的ACGT含量 |
| FTC | ACGT raw counters for first fragments | Read1片段, ACGT的原始计数 |
| LBC | ACGT content per cycle for last fragments only | Read2片段每个cycle的ACGT含量 |
| LTC | ACGT raw counters for last fragments | Read2片段, ACGT的原始计数 |
| BCC | ACGT content per cycle for BC barcode | BC Barcode每个周期的ACGT含量 |
| CRC | ACGT content per cycle for CR barcode | CR Barcode每个周期的ACGT含量 |
| OXC | ACGT content per cycle for OX barcode | OX Barcode每个周期的ACGT含量 |
| RXC | ACGT content per cycle for RX barcode | RX Barcode每个周期的ACGT内容 |
| QTQ | Quality distribution for BC barcode | BC Barcod的质量分布 |
| CYQ | Quality distribution for CR barcode | CR Barcod的质量分布 |
| BZQ | Quality distribution for OX barcode | OX Barcod的质量分布 |
| QXQ | Quality distribution for RX barcode | RX Barcod的质量分布 |
| IS | Insert sizes | 插入片段大小 |
| RL | Read lengths | read长度 |
| FRL | Read lengths for first fragments only | 只有Read1的长度 |
| LRL | Read lengths for last fragments only | 只有Read2的长度 |
| ID | Indel size distribution | Indel大小分布 |
| IC | Indels per cycle | 每个周期的Indels |
| COV | Coverage (depth) distribution | 覆盖范围(深度)分布 |
| GCD | GC-depth | GC深度 |
SN关键字信息说明(使用 grep ^SN | cut -f 2- 提取信息): samtools版本v1.6
| Key | Value | NOTE | idx | 说明 |
|---|---|---|---|---|
| raw total sequences: | 7693458 | (1) | 原始序列数 | |
| filtered sequences: | 0 | (2) | 过滤掉的序列数 | |
| sequences: | 7693458 | (3) | 序列数, 同(1) | |
| is sorted: | 1 | (4) | 是否排序, 1是,0否 | |
| 1st fragments: | 3876581 | (5) | R1片段数 | |
| last fragments: | 3816877 | (6) | R2片段数 | |
| reads mapped: | 6590466 | (7) | 比对上的reads数 | |
| reads mapped and paired: | 5329118 | # paired-end technology bit set + both mates mapped | (8) | 比对上,且是成对的reads数 |
| reads unmapped: | 1102992 | (9) | 未比对上的reads数 | |
| reads properly paired: | 4719116 | # proper-pair bit set | (10) | 成对比上也位置合适的序列(比对的位置是接近的?) |
| reads paired: | 7693458 | # paired-end technology bit set | (11) | 成对的read数(技术上) |
| reads duplicated: | 0 | # PCR or optical duplicate bit set | (12) | PCR或光学重复reads数 |
| reads MQ0: | 63803 | # mapped and MQ=0 | (13) | 比对质量是0的reads数 |
| reads QC failed: | 0 | (14) | 质量失败的reads数 | |
| non-primary alignments: | 266109 | (15) | 非首次比对的reads数 | |
| total length: | 1111705142 | # ignores clipping | (16) | 总长度(忽略截断) |
| bases mapped: | 952346426 | # ignores clipping | (17) | 比对上的碱基数(忽略截断) |
| bases mapped (cigar): | 921111215 | # more accurate | (18) | 来自Cigar的比对上的碱基数(更精确) |
| bases trimmed: | 0 | (19) | 去掉的碱基数 | |
| bases duplicated: | 0 | (20) | 重复的碱基数 | |
| mismatches: | 1822453 | # from NM fields | (21) | 错配数(来自NM) |
| error rate: | 1.98E-03 | # mismatches / bases mapped (cigar) | (22) | 错误率(错配数/比对上的总碱基数) |
| average length: | 144 | (23) | 平均长度 | |
| maximum length: | 150 | (24) | 最大长度 | |
| average quality: | 36.1 | (25) | 平均碱基质量 | |
| insert size average: | 845.9 | (26) | 插入片段长度 | |
| insert size standard deviation: | 1597.6 | (27) | 插入片段长度标准差 | |
| inward oriented pairs: | 2014285 | (28) | ?向内成对 | |
| outward oriented pairs: | 72676 | (29) | ?向外成对 | |
| pairs with other orientation: | 178825 | (30) | 其他方向 | |
| pairs on different chromosomes: | 2562 | (31) | 成对,比对在不同染色体上 |
上述示例信息是使用samtools v1.6 统计,发现与samtools v1.2结果有差异。
samtools flagstat统计使用v1.6和v1.2没有差异:
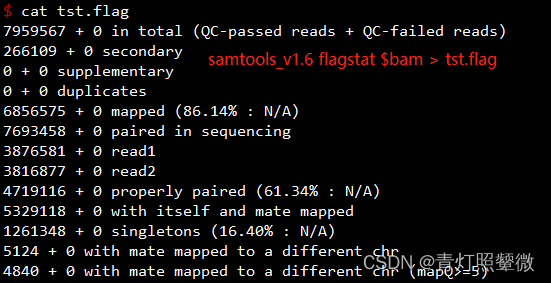
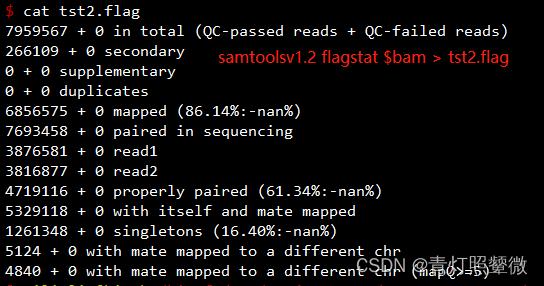
| key | 对应SN |
|---|---|
| 0 in total (QC-passed reads + QC-failed reads) | v1.2 (1) |
| 0 secondary | v1.2/v1.6 (15) |
| 0 supplementary | 补充比对reads数 |
| 0 duplicates | v1.2/v1.6 (12) |
| 0 mapped (86.14% : N/A) | v1.2 (7) |
| 0 paired in sequencing | v1.6 (1) |
| 0 read1 | v1.6 (5) |
| 0 read2 | v1.6 (6) |
| 0 properly paired (61.34% : N/A) | v1.6 (10) |
| 0 with itself and mate mapped | v1.6 (8) |
| 0 singletons (16.40% : N/A) | ? |
| 0 with mate mapped to a different chr | v1.6 (31) * 2 |
| 0 with mate mapped to a different chr (mapQ>=5) | 比对到不同染色体且比对质量值>5 |
一些问题:
【1】输入的bam/sam文件是PE,SN中的raw total sequences为什么是奇数,R1和R2的总和应该是偶数?
【2】官网samtools stats中对raw total sequences解释是:

就是说,统计的是:输入文件中 除了supplementary和secondary比对的reads,其他read数目。并且和samtools这个命令输出的是相同的:samtools view -c -F 0x900。
但是,在用samtools v1.2测试时,输出的raw total sequences的值和这个命令输出的reads数不同。且经过使用脚本自己统计read数,使用这个命令(samtools view -c -F 0x900)的结果是一致的。
import pysam
def cntbam(bam):
total_cnt = 0 # total read
bmrd = pysam.AlignmentFile(bam)
for rec in bmrd:
if rec.is_unmapped:
total_cnt += 1
continue
if 'H' in rec.cigarstring:
continue
if rec.is_secondary:
continue
if rec.is_supplementary:
print('#sup\t%s' % rec.query_name)
continue
total_cnt += 1
return total_cnt
print(cntbam(inbam))
pysam常用属性可参考笔者的另一篇博客:使用pysam读取sam/vcf/fasta文件时的常用属性

















 7132
7132

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









