







 本文介绍了samtools的三个重要命令:idxstats用于查看bam文件的比对统计信息,bedcov用于计算BED文件指定区域的碱基覆盖,faidx则用于对fasta文件建立索引及提取子序列。详细阐述了各命令的用法及输出结果解析。
本文介绍了samtools的三个重要命令:idxstats用于查看bam文件的比对统计信息,bedcov用于计算BED文件指定区域的碱基覆盖,faidx则用于对fasta文件建立索引及提取子序列。详细阐述了各命令的用法及输出结果解析。
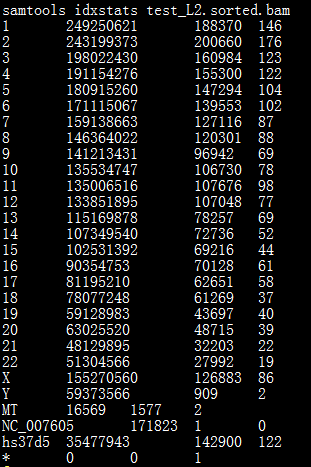
samtools idxstats命令功能简介:
检索和打印已建立索引的bam文件的统计数据,包括参考序列名称、序列长度、比对上的read数量和未比对上的read数量。输出结果显示在屏幕上,以制表符分割。
命令格式:
samtools idxstats <in.bam>
如下图所示:
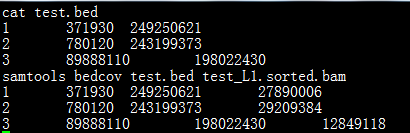
samtools bedcov命令功能简介:
计算由BED文件指定的基因组区域内的总碱基数量。
命令格式:
samtools bedcovregion.bed <in.bam | in.sam | in.cram>
如下图所示:
samtools faidx命令功能简介:
对fasta格式的参考序列建立索引或者从已经创建索引的参考序列中提取一段序列。如果没有指定区域,faidx命令就创建文件索引并生成后缀为.fai的索引文件。如果指定区域,那么就是生产并显示fasta格式的子序列。输入文件可以使BGZF压缩格式的文件。
另外,输入文件中的序列要有不同的名称。如果不是这样,即存在相同名称的序列,在建立索引的过程中将发出有关重复序列的警告而且生产的同名子序列的信息都要被第一个同名子序列的信息覆盖。
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 932
932

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









