






 浙江大学、中南大学和华东理工大学的研究者合作提出RELATION模型,利用3D结合构象和双向迁移学习设计新药。模型能生成具有高亲和力和药效团的分子,成功应用于AKT1和CDK2抑制剂设计,展示了在药物发现中的高效性和特异性。
浙江大学、中南大学和华东理工大学的研究者合作提出RELATION模型,利用3D结合构象和双向迁移学习设计新药。模型能生成具有高亲和力和药效团的分子,成功应用于AKT1和CDK2抑制剂设计,展示了在药物发现中的高效性和特异性。

这次给大家介绍一篇来自浙江大学侯廷军教授课题组、中南大学曹东升教授课题组、华东理工大学李洪林教授课题组联合发表在Journal of Medicinal Chemistry的《RELATION: A Deep Generative Model for Structure-Based De Novo Drug Design》,文章提出了一种基于蛋白质-配体复合物的3D结合构象的分子生成模型RELATION,该模型采用DSN架构来促进双向迁移学习(TL)中配体和配体-蛋白质复合物之间的信息交换,可以有效地生成具有良好结合亲和力和药效团特征的新分子。
摘要
基于深度学习(DL)的从头分子设计最近得到了大量关注。许多基于DL的生成模型已经成功应用于新分子设计,但其中大多数是以配体中心的,靶标结合蛋白质口袋的3D几何形状在分子生成中的作用尚未得到很好的利用。本文提出了一种基于3D的生成模型,称为RELATION。在RELATION模型中,BiTL算法专门用于提取蛋白质-配体复合物的所需几何特征并将其转移到潜在空间进行分子生成。药效团调节和基于对接的贝叶斯采样被应用于广阔化学空间的导航,以设计具有所需几何性质和药效团特征的分子。作为概念验证,使用RELATION模型设计了AKT1和CDK2两个靶点的抑制剂。计算结果表明,RELATION模型可以有效地生成具有良好亲和力和药效团特征的新分子。
1.研究背景
药物发现的主要目标之一是鉴定具有所需药理特性的新型化学实体。在药物发现早期鉴定出的候选药物在后期临床试验中失败的风险很高(约95%),导致资源浪费。从头药物设计旨在通过利用先进的计算技术生成具有新颖结构的潜在候选药物来加速药物开发并降低成本。通常,传统的从头药物设计方法可以分为两类:基于配体的和基于结构的。经典的基于配体的方法,需要一组经过实验验证的活性化合物作为起点,活性化合物共享的共同特征可以随后被利用来指导新型候选药物的设计。基于结构的方法,通过递增地添加、删除、插入或替换嵌入目标结合口袋中的化学支架的片段来创造新的小分子。
基于深度学习的一些先进的生成算法已被用于新药设计,如递归神经网络、编码−解码器(Enc-Dec)、生成对抗网络(GAN)和强化学习(RL)等。但大多数基于DL的生成算法都是以配体为中心的。因此,分子被表示为1D的SMILES串或2D的分子图。一维/二维表示的一个明显缺点是生成模型无法捕获3D分子几何形状对配体结合的关键影响,可能不足以用于基于结构的从头药物设计中的目标特异性任务。有一部分模型是通过将配体或/和蛋白质的3D特征纳入基于DL的生成架构而开发的,但这部分模型没有同时解决从头药物设计中的关键点,如配体结合模式和生成分子的药效团特征。
作者提出了RELATION(REceptor-LigAnd interacTION),这是一种基于Enc-Dec的从头药物设计生成模型。与现有的DL方法不同,模型以具有原子物理化学性质的配体−受体复合体的三维网格构象为输入,因此,它在生成具有良好的药效团特征和结合模式的特定靶点的分子方面明显更高效。采用结构域分离网络(DSN)进行双向迁移学习(TL),来促进配体和配体−蛋白复合物之间的信息交换。为了不断提高模型的有效性,药效团的约束和基于对接分数的抽样分别由条件生成和对接分数抽样获得。最后,利用该模型设计了AKT1和CDK2的潜在抑制剂。
2.RELATION模型
2.1 数据集准备
由于RELATION引入了双向TL,所以模型训练使用了源域和目标域两种数据集,其中源域的数据集来源于ZINC数据库,除碳、氮、氧、硫、氟、氯、溴和氢之外,还含有带电原子或原子的分子被去除,选择分子量在200到600之间,logP(通过RDKit中的Crippen模型计算)在−2到6之间的分子,共包含1,200,000个小分子;从BindingDB、ChEMBL和PDB数据库中收集了407种AKT1抑制剂和1017种CDK2抑制剂(IC50< 50 nM),然后将两个靶点的抑制剂对接到靶标蛋白,只保留配体周围5 Å的原子作为蛋白配体复合物数据集。最后,源数据集和目标数据集中的每个分子由其坐标(张量的前三个维度)和特征向量定义的4D张量表示。
2.2 模型架构
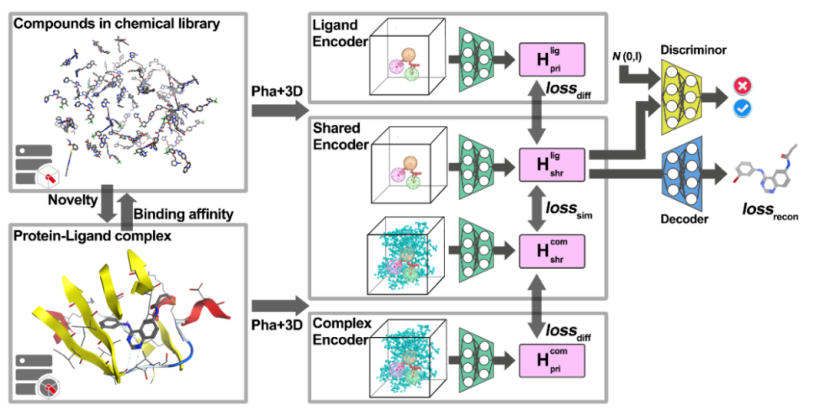
图1.RELATION模型架构图
基于VAE的RELATION模型主要由两个部分组成,如图1所示。第一部分是3D卷积编码器,包括私有编码器和共享编码器,第二部分是caption-LSTM解码器。训练数据转换为4D张量后,进入编码器通过八个隐藏层。第一层包含64个滤波器,然后在每个奇数层上加倍,最后一层包含512个滤波器。每个偶数层后面都有一个额外的池化层,内核大小、步幅和填充为2,以执行下采样。利用ReLU激活函数对3D-CNN模型进行训练,生成并合并一个1024维的嵌入向量。caption-LSTM解码器可以将隐藏层内的高维向量转化为SMILE分子式,caption-LSTM由三层组成,其词汇量输入大小为39,隐藏大小为1024。
作者还试图通过将GAN框架引入架构来探索基于AAE的关系模型的有效性,在基于AAE的RELATION模型中,添加了一个额外的判别器,输入数据通过共享和私有编码器转换为潜码H,然后由解码器网络解码。另一方面,判别器将预测给定向量是由AAE生成的还是从N(0,1)中采样的向量。
2.3 RELATION的损失函数
受DSN的启发,总损失函数定义如下:
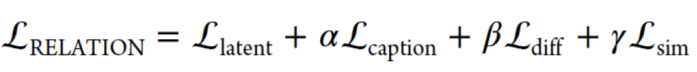
Lsim和βLdiff的引入使隐藏层能够同时使用源数据集和目标数据集进行双向TL。其中,α,β和γ表示控制每个损失项的权重因数,并且分别被设置为0.01、0.075和0.25,Llantent表示编码器被先验正则化,先验遵循均值和单位方差为零的多元高斯分布,Lcaption为重构损失函数,Ldiff用于保证隐藏层小分子和复合物各自数据的特点,Lsim用于评估配体和复杂数据集之间的相似性。
3.实验与结果
3.1 生成分子的性质和质量
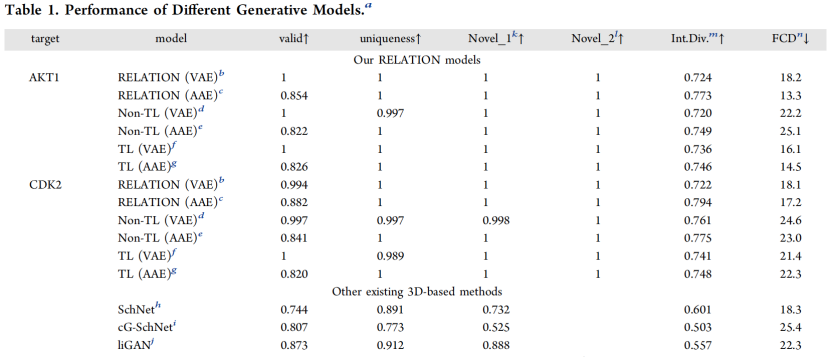
表1.不同生成模型的表现(其中,Novel_1:是指在源数据集中和其他现有数据集中找不到的生成化合物的比例;Novel_2:是指生成的化合物中没有在现有抑制剂数据集中找到的比例;mInt.Div.:表示内部多样性)
从表1中可以看到,可以发现具有双向TL的RELATION(AAE)和RELATION(VAE)模型比其他模型产生了更高的有效性、唯一性和内部多样性分数。FCD(Fréchet ChemNet距离)值可用于评估生成分子与参考数据集之间化学和生物性质的相似性。显然,TL模型产生的FCD值比非TL模型相对较低,表明使用TL技术时生成分子的化学和生物性质与现有抑制剂更相似。
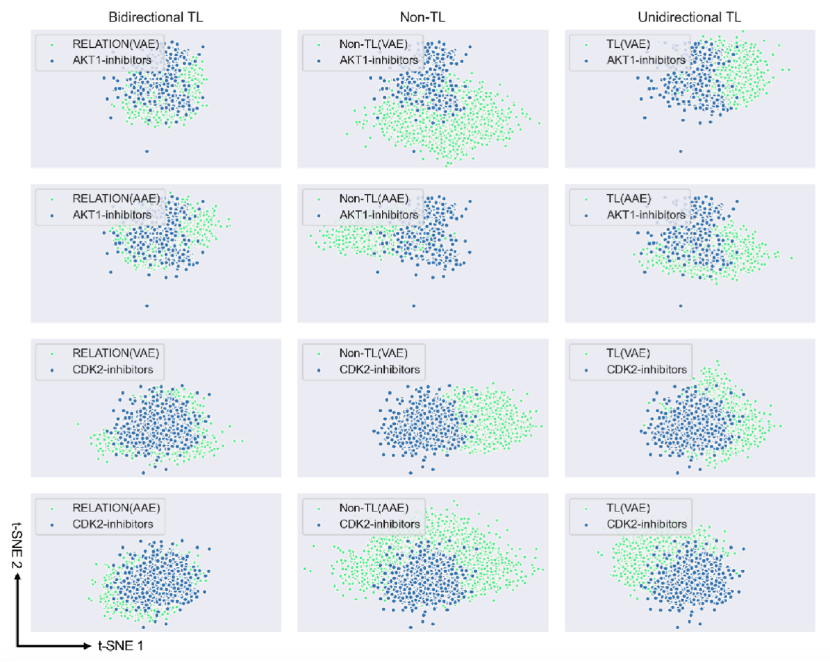
图2. 不同模型和现有抑制剂产生的10,000个有效分子的T-SNE图(蓝色点指的是AKT1和CDK2抑制剂,绿色点代表不同模型产生的分子)
根据图2中的非TL列可以看出,与现有抑制剂相比,VAE和对抗自编码器(AAE)生成的分子分布在完全不同的空间,当使用单向TL算法重新训练模型时,生成的分子分布与现有抑制剂之间的重叠明显增加。如图2中的双向TL列所示,RELATION(VAE)和RELATION(AAE)生成的分子几乎完全与现有抑制剂重叠,表明通过在VAE和AAE架构上使用双向TL,生成的分子和现有抑制剂覆盖了相似的化学空间。这些结果也与表1所示的数据(FCD值)一致。因此,随着双向TL算法和几何构象的应用,作者提出的RELATION模型可以捕获训练数据的潜在特征,RELATION模型在生成分子的有效性、唯一性、新颖性和多样性等方面的性能优于其他现有的基于3D的生成模型。
3.2 输入方向的结果的稳定性
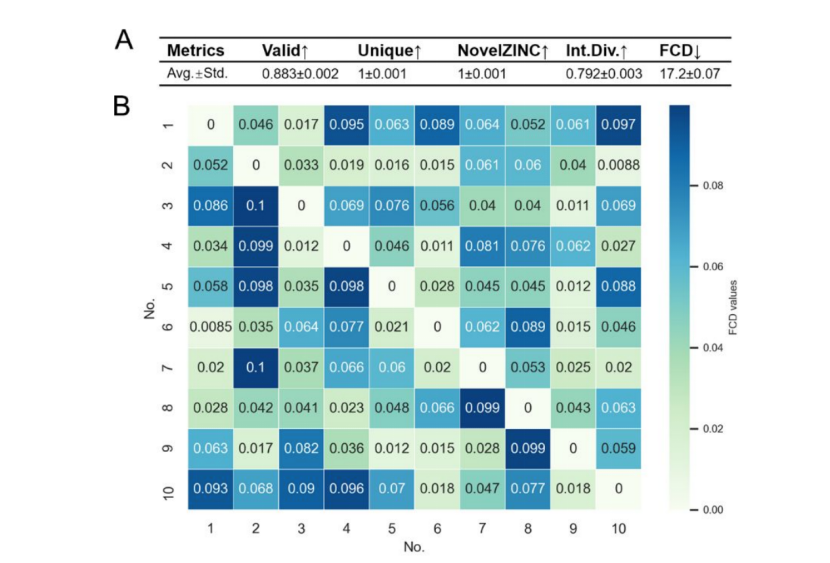
图3.(A)用不同方向的输入数据集训练的10个关系模型生成的分子集的度量(B)热图表示10个分子集中每对的FCD值
作者使用了10个具有不同方向的AKT1源数据集-目标数据集(在生成分子的质量和属性部分中提到)来训练关系(AAE)模型。最终结果可以发现生成的分子集的质量没有表现出明显的变化。
3.3 RELATION和基于药效团的RELATION的比较
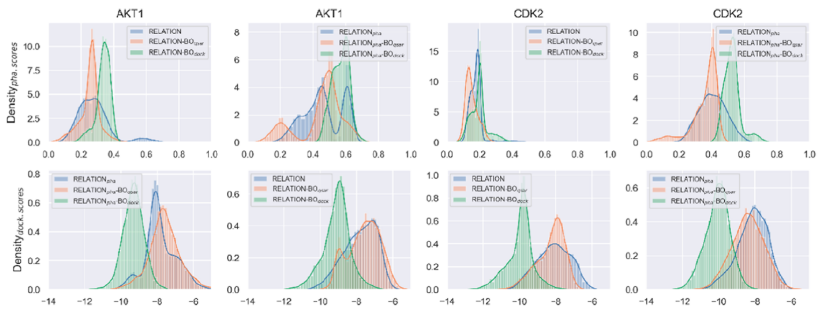
图4.不同模型生成的10000个有效分子的KDE(内核密度估计)分布
作者进一步将条件VAE(CVAE)中的药效团特征引入到RELATION的训练中,以使生成过程更适合目标特异性任务。对比的RELATION是指基于AAE的RELATION(因为它在FCD指标方面的表现优于基于VAE的RELATION)。生成分子的药效团分数(rel.SFCR)的分布如图4所示。对于AKT1和CDK2,RELATIONpha(基于药效团的RELATION模型)生成的分子比原始RELATION模型生成的分子具有更高的药效团分数。这表明通过将药效团特征引入RELATION,生成的分子可以增强与预设药效团模型的匹配。
3.4 RELATION模型的贝叶斯优化
为了提高RELATIONpha模型的采样效率,并在显著较大的化学空间中生成一组具有良好对接构象的有效分子,作者还在模型中进入了贝叶斯优化(Bayesian Optimization,BO)的采样,如图4所示,生成分子的药效团分数得到了提高,其中BOdock的性能略优于BOqsar。BOdock生成的分子的对接分数与原始RELATION模型生成的分子相比有显著提高,但BOqsar生成的分子的QSAR分数仅略有变化。还发现,BOdock生成的分子表现出与现有抑制剂相似的药效团和对接分数分布。这些结果表明,BOdock产生的分子更有可能同时具有良好的药效团和对接特征,因此,BOdock可能是靶向特异性任务的更好选择。
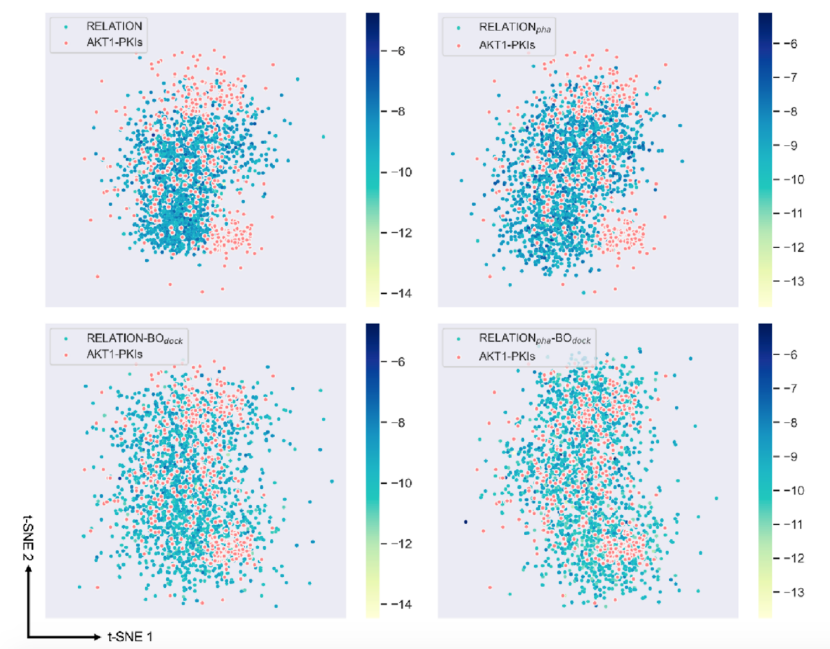
图5.RELATION模型使用不同的采样方式生成分子的化学空间分布
为了进一步研究基于BO采样的RELATION模型的性能,作者将不同模型生成的有效分子与AKT1抑制剂再次进行了T-SNE分析。如图5所示,RELATION和RELATIONpha模型不能有效地探索AKT1抑制剂的化学空间(红色圆圈中标记的点)。然而,将BO采样引入RELATION模型后,采样分子在化学空间中的分布比原始RELATION模型更加分散,这表明生成分子的与AKT1空间更相似。此外,根据点的颜色梯度,从基于BO的RELATION模型中采样的分子比从原始RELATION模型中采样的分子表现出更有利的对接分数。
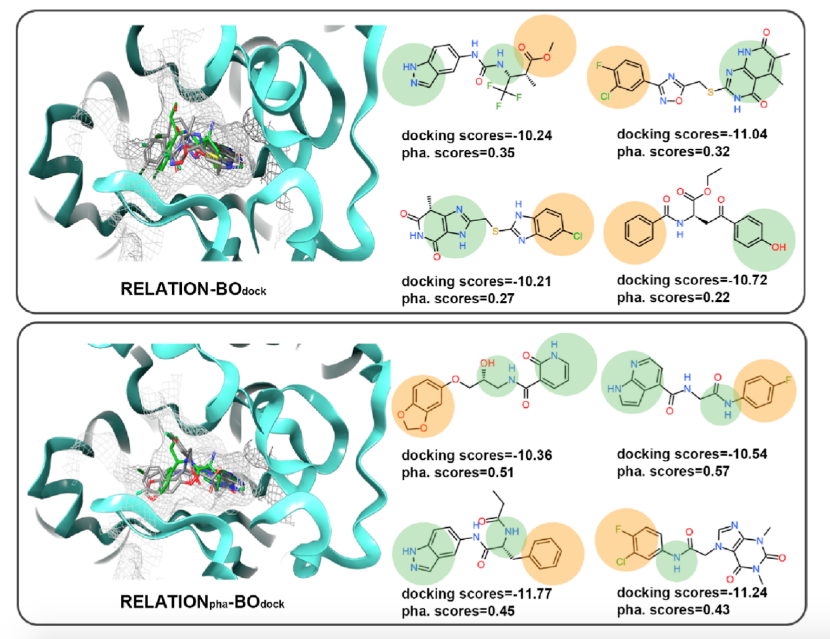
图6.来自RELATION-BOdock和RELATIONpha-BOdock的采样分子
如图6所示,可以发现生成的分子与AKT1抑制剂构象相同,朝向铰链区、酸孔和环区的方向相同,右侧的药效团特征也与理想的AKT1抑制剂一致。同时,对接和药效团评分结果表明,随着BO采样的引入,RELATION和RELATIONpha都能产生具有良好对接分数的分子,但BO-RELATIONpha产生的分子具有更高的药效团匹配分数。而且,通过将药效团特征引入RELATION架构,模型可以根据预设的药效团特征进行条件生成。
4.结论
本文提出的RELATION模型,可以生成新颖、有效、多样且亲和力高的分子。其中引入的双向TL在模拟现有抑制剂的理化性质方面比其他方法有更大的优势;为了能够创建具有所需特征的新分子,引入了药效团约束和基于BO的采样来优化RELATION,优化后的模型生成的分子与现有抑制剂具有更好的结合亲和力和更高的相似性,并具有更高的药效团匹配分数。总体来看,本文提出的RELATION模型在这些目标特异性任务中有效性较高。是一个基于结构的从头药物设计的有力工具。
参考资料:
Wang M, Hsieh C Y, Wang J, et al. RELATION: A Deep Generative Model for Structure-Based De Novo Drug Design[J]. Journal of Medicinal Chemistry, 2022, 65(13): 9478-9492.
https://doi.org/10.1021/acs.jmedchem.2c00732

















 2416
2416

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









