







 AUCell是一种用于分析基因集在单细胞表达数据中富集程度的方法,通过计算AUC分数来评估特征在不同细胞中的相对表达。文章介绍了AUCell的使用场景、安装包依赖、步骤以及一个示例代码,展示了如何利用AUCell进行单细胞转录组数据分析和可视化。
AUCell是一种用于分析基因集在单细胞表达数据中富集程度的方法,通过计算AUC分数来评估特征在不同细胞中的相对表达。文章介绍了AUCell的使用场景、安装包依赖、步骤以及一个示例代码,展示了如何利用AUCell进行单细胞转录组数据分析和可视化。
AUCell介绍
AUCell是什么:AUCell使用曲线下面积来计算输入基因集的一个有意义的基因子集是否在每个细胞的表达基因中富集。AUC 分数在所有细胞中的分布允许探索特征的相对表达。由于评分方法是基于排名的,因此 AUCell 与基因表达单位和归一化程序无关。此外,由于细胞是单独评估的,因此可以很容易地应用于更大的数据集。
AUCell的使用场景很灵活,例如当我们找到一组关键的差异表达基因集合,可能从文献的补充材料中下载的,或者公共数据库中收集的,在基因数超过100,甚至1000时,通过dotplot和umap染色图一个个看过于主观且工作量大,这种情况下,总和考虑所有基因集的表达特征,将他们映射到整个转录组数据上,可以明确的发现该基因集在哪些细胞中高富集。
AUCell安装
if (!requireNamespace("BiocManager", quietly=TRUE))
install.packages("BiocManager")
# To support paralell execution:
BiocManager::install(c("doMC", "doRNG","doSNOW"))
# For the main example:
BiocManager::install(c("mixtools", "SummarizedExperiment"))
# For the examples in the follow-up section of the tutorial:
BiocManager::install(c("DT", "plotly", "NMF", "d3heatmap", "shiny", "rbokeh",
"dynamicTreeCut","R2HTML","Rtsne", "zoo"))AUCell步骤
Step 1: 根据基因表达量计算每个细胞中每个基因的排名
cells_rankings <- AUCell_buildRankings(exprMatrix)
dataframe <- data.frame(cells_rankings@assays@data$ranking)
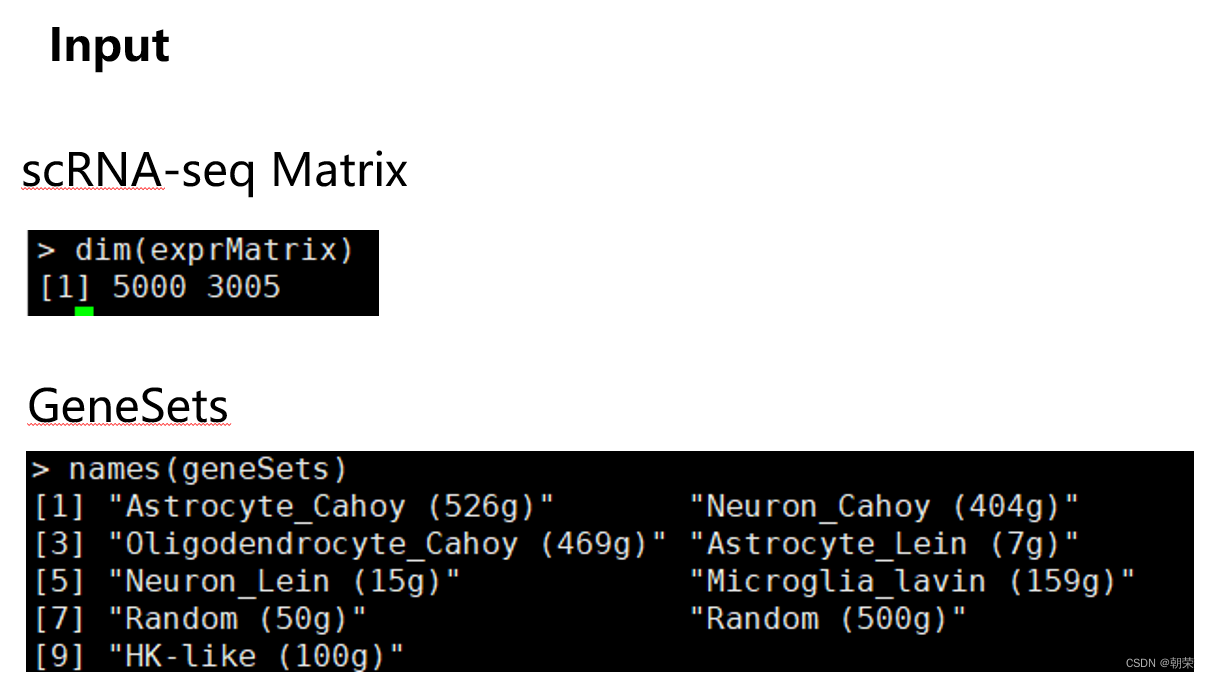
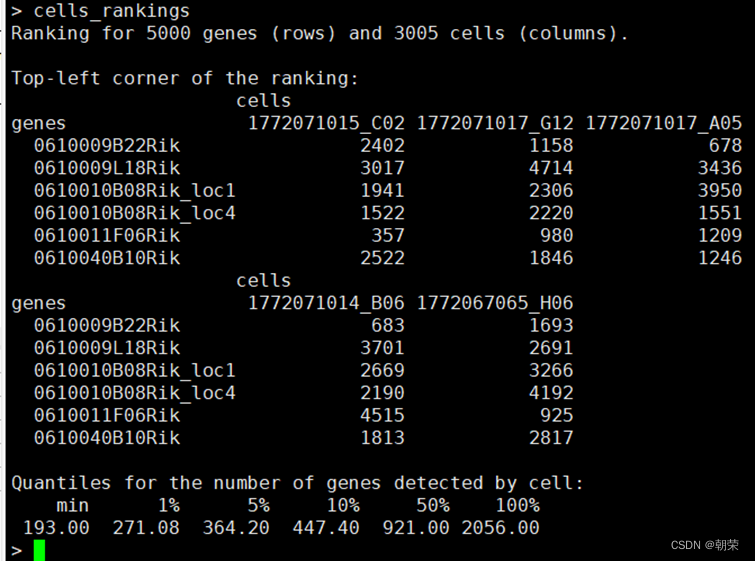
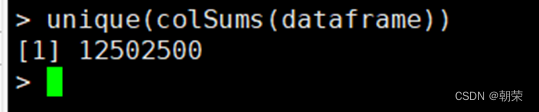
统计了每一列排名的总和,确实为1~5000,对于相同的UMI counts,随机排名
Step 2: 计算每个细胞中每个基因集的AUCell分数(5% of total genes)(可选范围:1%~20%,对于每个cell中表达丰富的单细胞数据可以适当提升阈值)
cells_AUC <- AUCell_calcAUC(geneSets, cells_rankings)
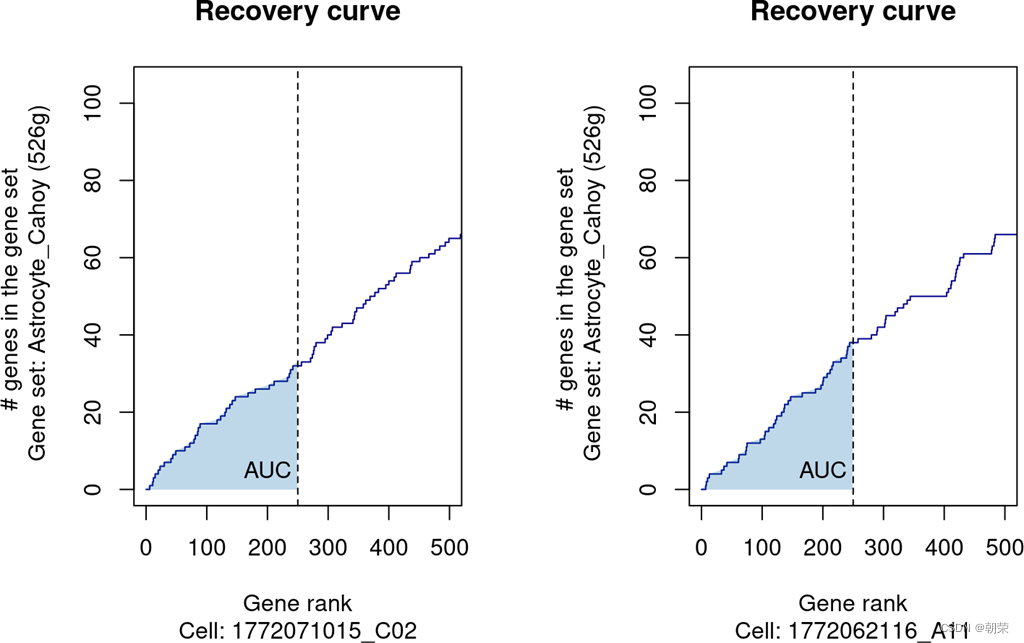
X轴表示前250个基因
Y轴表示对应x轴重top n个基因在GeneSet中的基因数量
当前GeneSet的AUCell分数:sum(阈值前x轴的差分 * y轴的差分) / x轴阈值 * GeneSet基因数
normAUC:参数决定是否将AUC最大值标准化为1 (Default: TRUE)

Step 3:设置阈值估计基因集中在每个细胞中高表达的基因的比例
AUCell的分布:最佳情况是双峰分布,数据集中的大多数细胞与具有明显较高值的细胞群相比具有较低的AUC
AuCell_exploreThresholds : Set the assignment thresholds
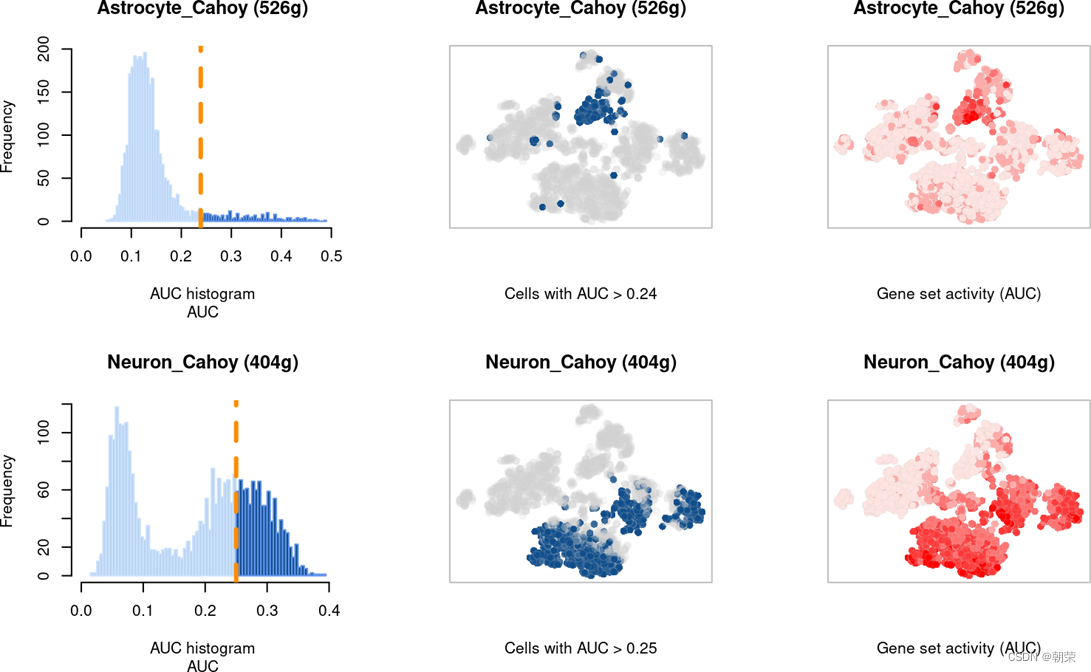
AUCell简易代码
library(AUCell) #可以加载成功即可
library(ggplot2)
library(patchwork)
library(dplyr)
library(Seurat)
#读取rds文件,并完成降维和聚类
rds_path = 'XXX/XXX/XX.rds'
rds <- readRDS(rds_path)
rds <- NormalizeData(rds)
rds <- FindVariableFeatures(rds, selection.method = "vst", nfeatures = 2000)
rds <- ScaleData(rds, verbose = FALSE)
rds <- RunPCA(rds, npcs = 100, seed.use = 42,verbose = FALSE)
rds <- RunUMAP(rds, reduction = "pca", dims = 1:60, seed.use =42)
#获得原始表达量作为matrix
matrix <- Seurat::GetAssayData(object=rds, slot="counts", assay="RNA")
set.seed(1)
#获取基因集,filtered_data 是一组基因名list
cell_type = "Root Cap"
filtered_data <- df[df$Celltype == cell_type, ]$geneID
#计算AUCell分数
cells_rankings <- AUCell_buildRankings(matrix, plotStats=FALSE)
cells_AUC <- AUCell_calcAUC(filtered_data, cells_rankings)
cells_assignment <- AUCell_exploreThresholds(cells_AUC, plotHist = TRUE, assign=TRUE)
thr = cells_assignment$geneSet$aucThr$selected;thr
new_cells <- names(which(getAUC(cells_AUC)["geneSet",]> thr))
#将结果保存在rds中
rds$is_list <- ifelse(colnames(rds) %in% new_cells, "list", "non_list")
#存储每种细胞类型中基因集激活的细胞数和基因集沉默的细胞数
tab <- table(rds$is_list,rds$Celltype)
write.table(tab, file = paste0(out_path,'/',cell_type,"_count.txt"), sep = "\t", quote = FALSE, col.names = NA)
#绘制umap图
p = DimPlot(object = rds, group.by = "is_list", label = TRUE) + DimPlot(object = rds, group.by = "Celltype", label = TRUE)
ggsave(p,file=paste0(out_path,'/',cell_type,"-list-UMAP.pdf"), width = 10, height = 5)
参考文献:Aibar et al. (2017) SCENIC: single-cell regulatory network inference and clustering. Nature Methods. doi: 10.1038/nmeth.4463

















 3810
3810

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









