







多组学分析R包汇总表
名称 | 用途 | 适用组学 | 功能介绍 | 输出形式 |
|---|---|---|---|---|
| mixOmics | 多组学数据整合与可视化 | 转录组、代谢组、蛋白质组、微生物组等 | 提供PLS、sPLS、DIABLO等方法进行多组学整合分析,支持降维和变量选择 | 交互式图表、变量权重图、网络图 |
| MOFA/MOFA+ | 多组学因子分析 | 任意组学组合(需矩阵格式) | 基于概率矩阵分解识别跨组学的潜在因子,处理缺失值和高维度数据 | 因子权重图、样本聚类图、热图 |
| omicade4 | 多组学联合分析 | 转录组、代谢组、蛋白质组等 | 基于多重协惯性分析(MCIA)进行多组学数据整合和可视化 | 联合排序图、变量贡献图 |
| IntegrOmics | 跨组学关联分析 | 基因组、转录组、蛋白质组等 | 提供统计方法整合不同组学层数据,寻找跨层关联关系 | 关联网络图、统计结果表 |
| MultiAssayExperiment | 多组学数据容器管理 | 任意组学组合 | 提供统一数据结构存储和同步管理多组学实验数据,支持与Bioconductor工具链集成 | 结构化数据对象 |
| CIMLR | 多组学聚类与分类 | 转录组、甲基化组、蛋白质组等 | 使用多核学习算法进行多组学聚类和样本分类,支持高维数据处理 | 聚类标签、分类概率、热图 |
| WGCNA | 加权基因共表达网络分析 | 转录组、代谢组、蛋白质组等 | 构建共表达网络识别模块,支持跨组学模块关联分析 | 网络图、模块特征表、相关性图 |
| omicsPCA | 多组学PCA整合分析 | 任意组学组合 | 扩展PCA方法实现多组学数据联合降维和可视化 | 联合PCA图、载荷矩阵 |
| iClusterPlus | 多组学亚型发现 | 基因组、转录组、表观组等 | 基于联合潜变量模型识别跨组学分子亚型 | 亚型分类、热图、生存曲线 |
| PMA | 惩罚多元分析 | 转录组、蛋白质组、代谢组等 | 实施稀疏CCA和回归方法寻找跨组学关联特征 | 特征权重表、相关性网络图 |
| NetICS | 多组学网络传播分析 | 基因组、转录组、蛋白质组等 | 基于网络传播算法整合多组学数据识别关键调控节点 | 节点评分、网络可视化 |
| omicsIntegrator | 多组学网络整合 | 蛋白质组、代谢组、相互作用组等 | 整合多组学数据构建分子相互作用网络,识别功能模块 | 网络文件、富集分析结果 |
简介
mixOmics是一个R包,用于探索和整合组学数据,包括转录组学,蛋白质组学,脂质组学,微生物组学,宏基因组学等。mixOmics软件包包括用于数据集成、生物标志物发现和数据可视化的工具,使用先进的多变量方法来降低数据维度并揭示数据集内部和数据集之间的关系。
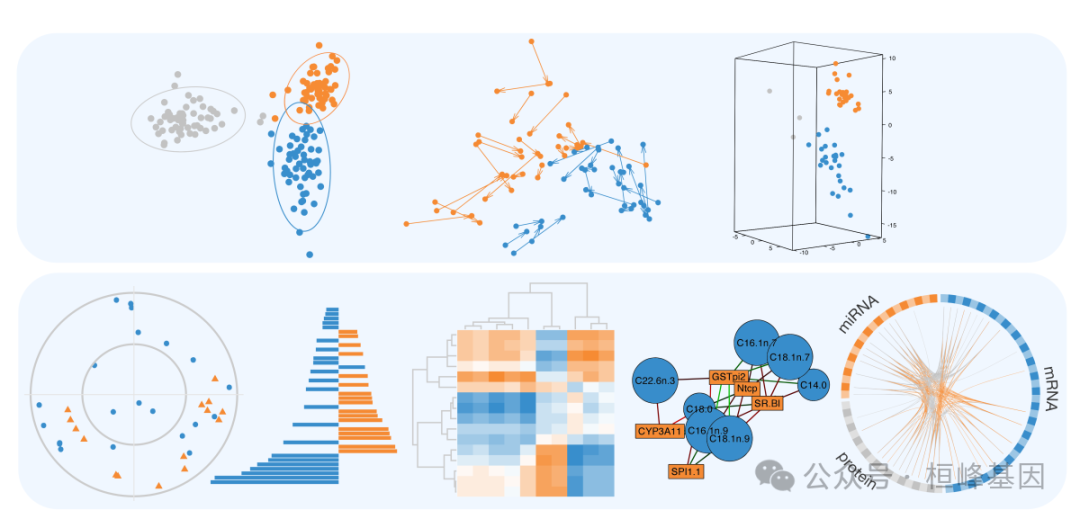
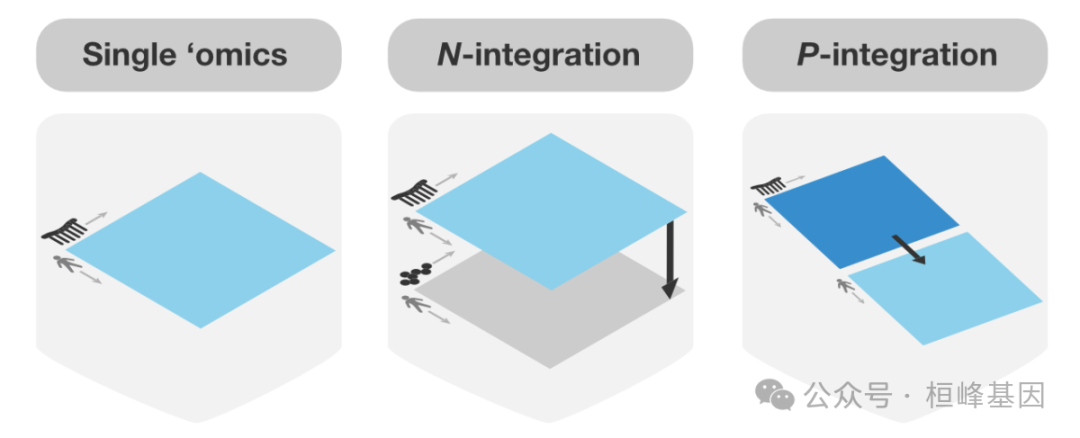
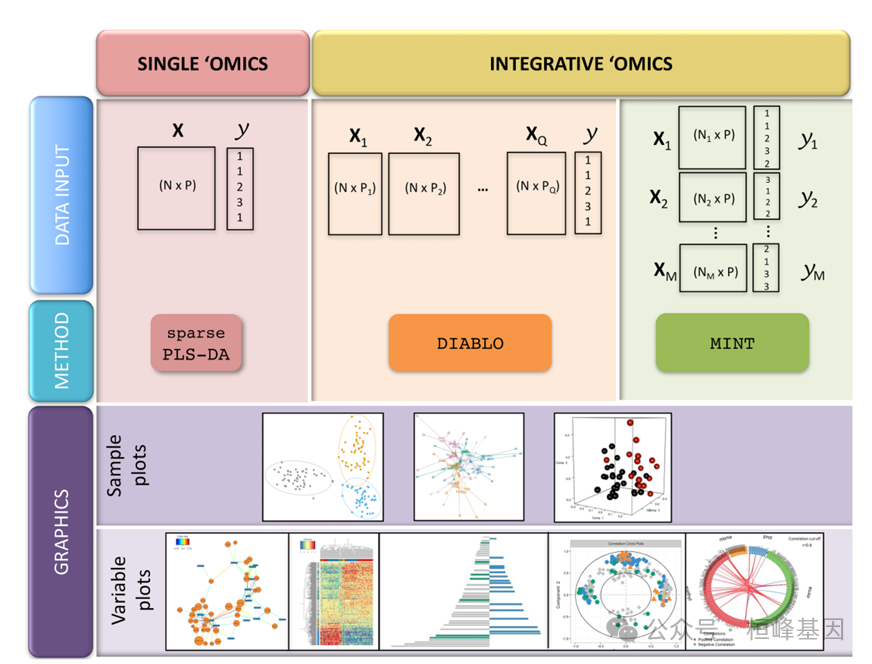
组学数据的多变量方法
多变量方法非常适合具有许多变量(例如,基因,蛋白质)和少量样本(例如,患者,细胞)的大型组学数据集。他们通过创建组件(变量的组合)来降低数据维度,从而揭示组学数据集之间的模式和关系。这些组件可用于产生清晰的可视化,揭示与生物学结果相关的关键变量,并集成多组学数据集。我们的稀疏多变量模型可用于识别高度相关的关键变量或解释感兴趣的生物学结果
安装与加载
从Bioconductor安装
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("mixOmics")加载包
library(mixOmics)数据准备
数据读取
utils::data(nutrimouse) # 内置小鼠营养研究数据集
#str(nutrimouse)
X <- nutrimouse$gene # 转录组数据 (40样本 x 120基因)
Y <- nutrimouse$lipid # 代谢组数据 (40样本 x 21脂质)
diet_group <- nutrimouse$diet # 饮食分组信息数据预处理
# 标准化处理
X_scaled <- scale(X)
Y_scaled <- scale(Y)例子实操
多组学整合分析 (sPLS方法)
# 参数调优选择变量数
tuned_model <- tune.spls(
X = X_scaled,
Y = Y_scaled,
ncomp = 5,
test.keepX = seq(5, 20, by = 5),
test.keepY = seq(3, 10, by = 2),
validation = "Mfold",
folds = 5,
nrepeat = 10
)# 查看最优参数
print(tuned_model$choice.keepX)## comp1 comp2 comp3 comp4 comp5
## 5 5 5 5 5print(tuned_model$choice.keepY)## comp1 comp2 comp3 comp4 comp5
## 3 9 3 7 7运行最终模型
final_model <- spls(X_scaled, Y_scaled,
ncomp = tuned_model$choice.ncomp+2,
keepX = tuned_model$choice.keepX,
keepY = tuned_model$choice.keepY)## keepX is of length 5 while ncomp is 3
## trimming keepX to [33mc(5,5,5)[39m结果可视化
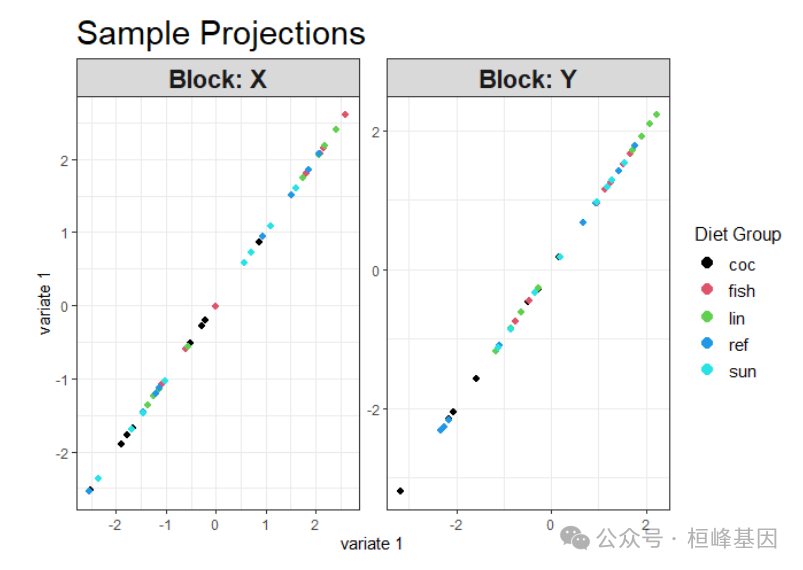
样本投影图 (带分组标签)
plotIndiv(
final_model,
comp = c(1,1),
group = diet_group,
pch = 16,
cex = 1.5,
col.per.group = 1:5,
legend = TRUE,
legend.title = "Diet Group",
title = "Sample Projections"
)
变量聚类图
plotVar(final_model)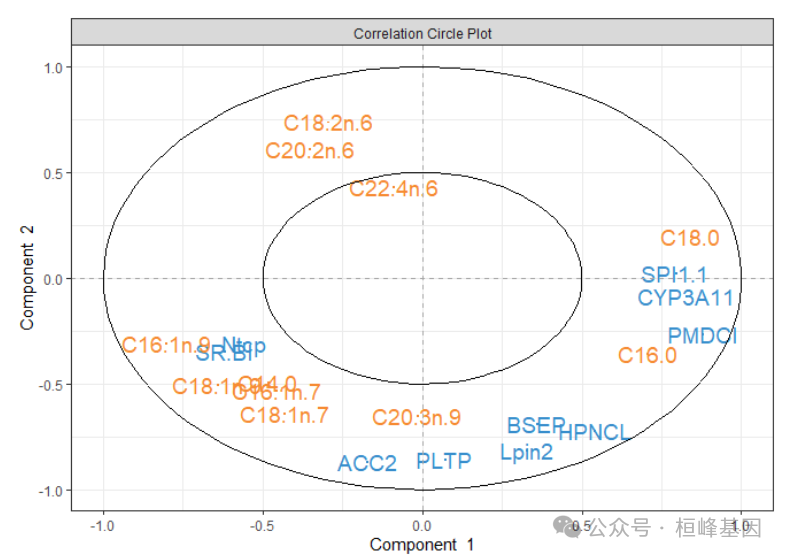
变量关联网络图
# 生成PDF报告
pdf("mixOmics_network.pdf", width=10, height=8)
network(
final_model,
comp = 1:2,
color.node = c("gene" = "#1F78B4", "lipid" = "#E31A1C"),
shape.node = c("gene" = "circle", "lipid" = "rectangle"),
alpha.node = 0.7,
cutoff = 0.6
)
dev.off()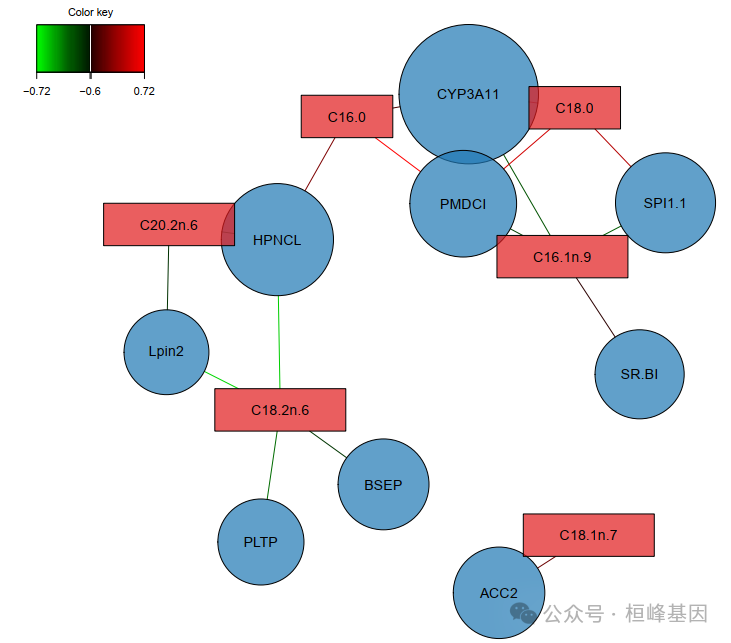
双标图
biplot(final_model,legend = TRUE,
legend.title = "Diet Group")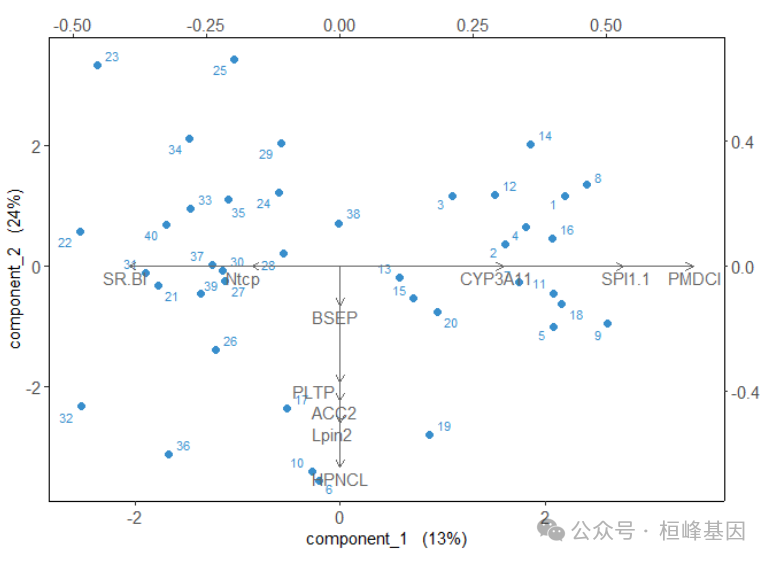
箭头图
plotArrow(final_model,legend = TRUE,
legend.title = "Diet Group")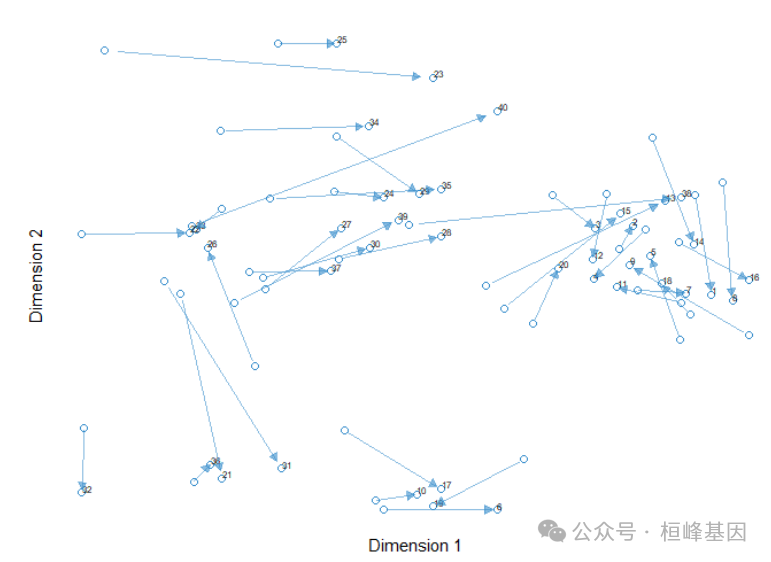
负载图
plotLoadings(final_model,legend = TRUE,
legend.title = "Diet Group")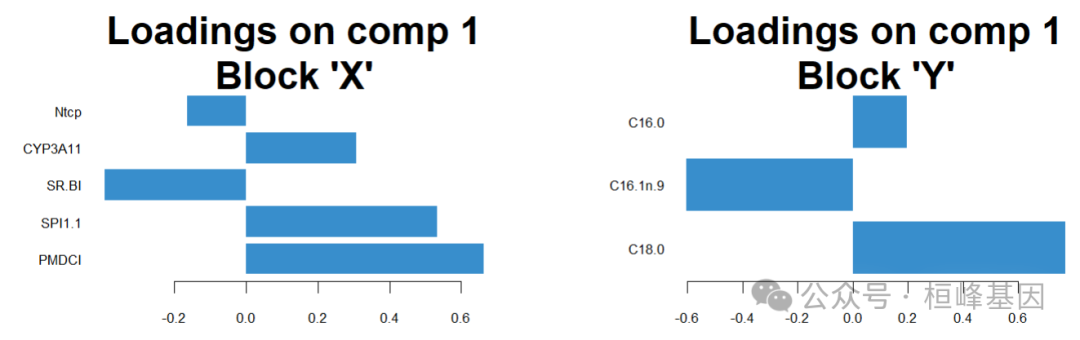
变量权重热图
jpeg(filename = "test1.jpeg", res = 600, width = 4000, height = 4000)
cim(cor(X, Y), cluster = "none")
dev.off()## CIM representation for objects of class 'rcc'
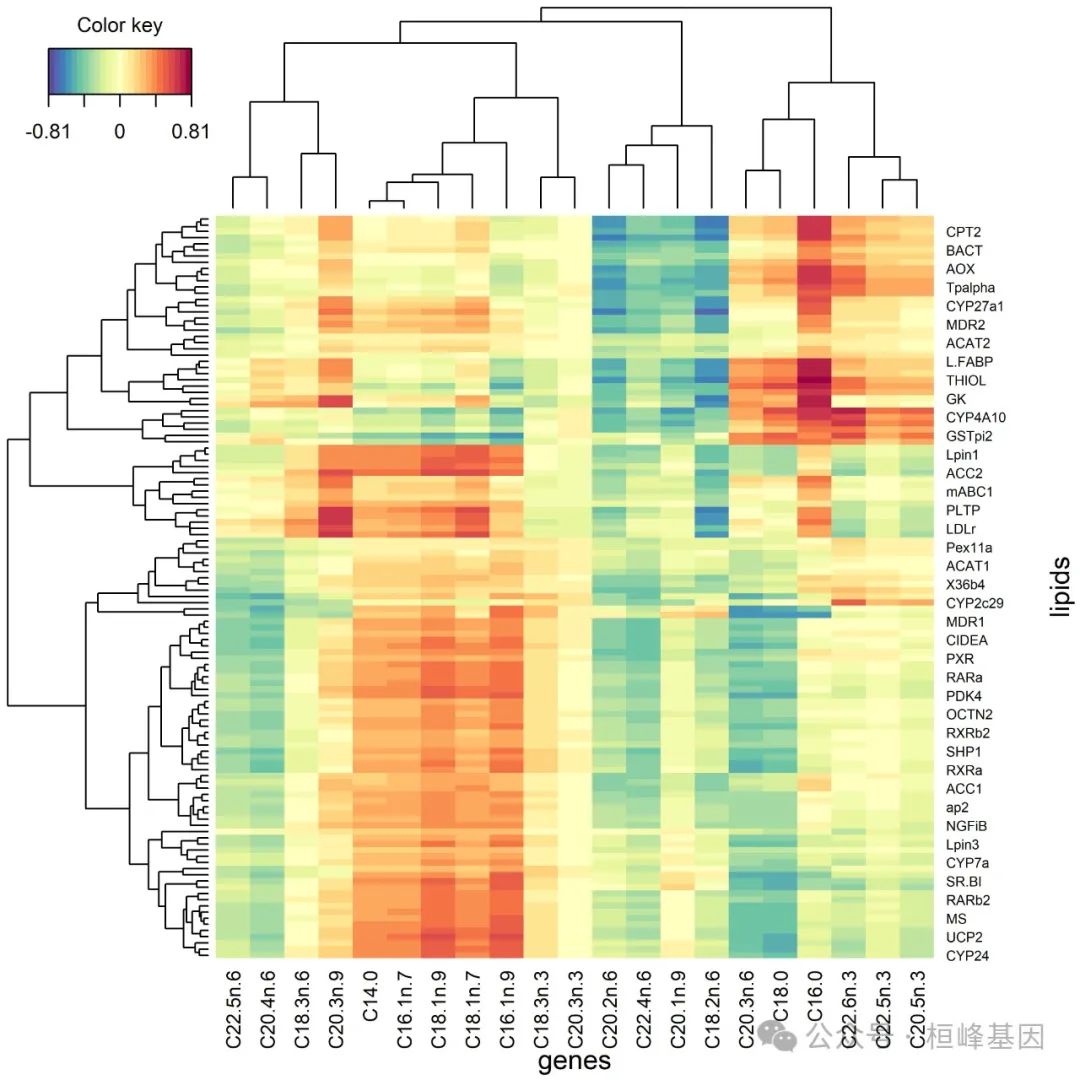
jpeg(filename = "test2.jpeg", res = 600, width = 4000, height = 4000)
nutri.rcc <- rcc(X, Y, ncomp = 3, lambda1 = 0.064, lambda2 = 0.008)
cim(nutri.rcc, xlab = "genes", ylab = "lipids", margins = c(5, 6))
dev.off()#-- cim from X matrix with a side bar to indicate the diet
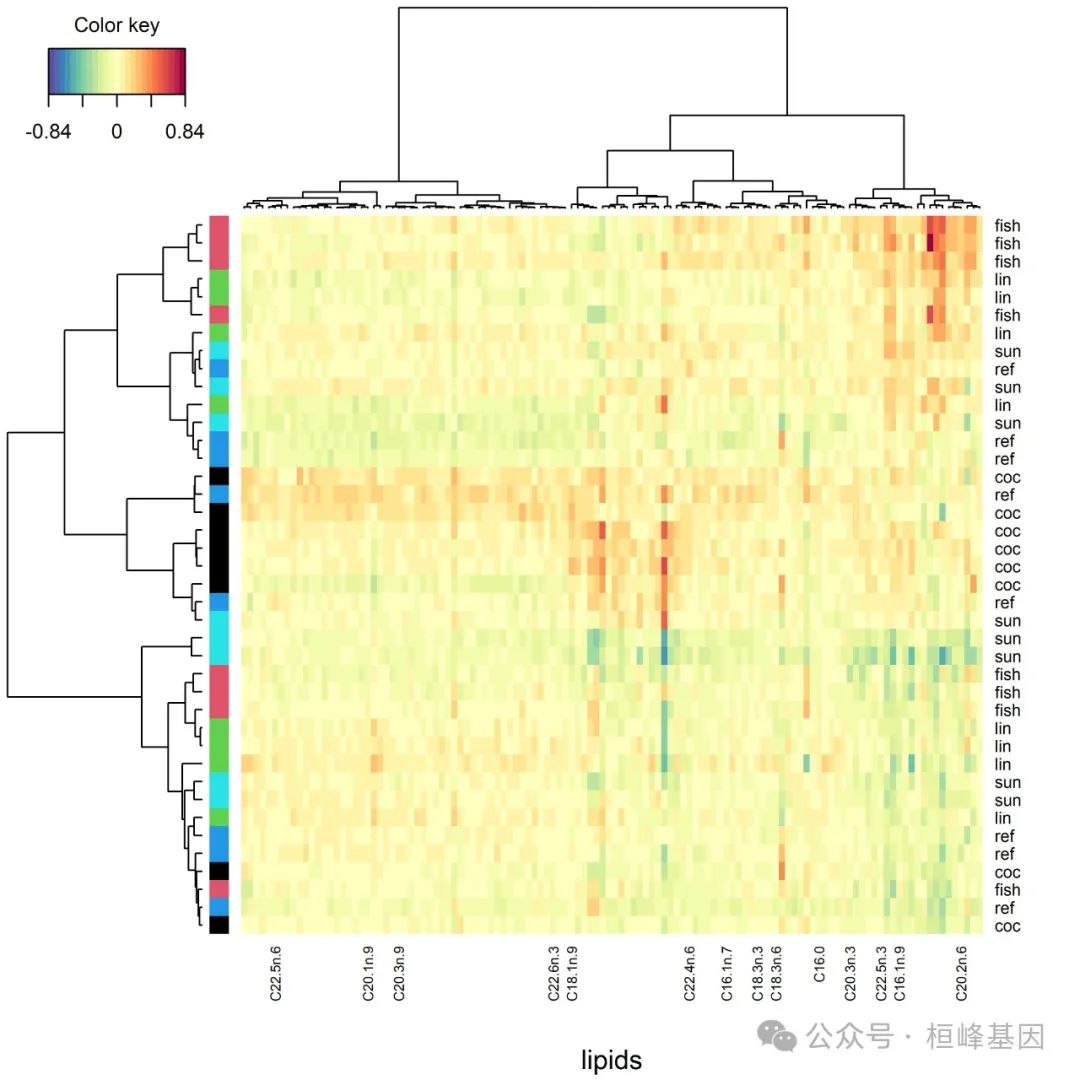
diet.col <- palette()[as.numeric(nutrimouse$diet)]
jpeg(filename = "test4.jpeg", res = 600, width = 4000, height = 4000)
cim(nutri.rcc, mapping = "X", row.names = nutrimouse$diet,
row.sideColors = diet.col, xlab = "lipids",
clust.method = c("ward", "ward"), margins = c(6, 4))
dev.off()#-- cim from Y matrix with a side bar to indicate the genotype
geno.col = color.mixo(as.numeric(nutrimouse$genotype))
jpeg(filename = "test5.jpeg", res = 600, width = 4000, height = 4000)
cim(nutri.rcc, mapping = "Y", row.names = nutrimouse$genotype,
row.sideColors = geno.col, xlab = "genes",
clust.method = c("ward", "ward"))
dev.off()jpeg(filename = "test6.jpeg", res = 600, width = 4000, height = 4000)
cim(nutri.rcc, xlab = "genes", ylab = "lipids", margins = c(5, 6))
dev.off()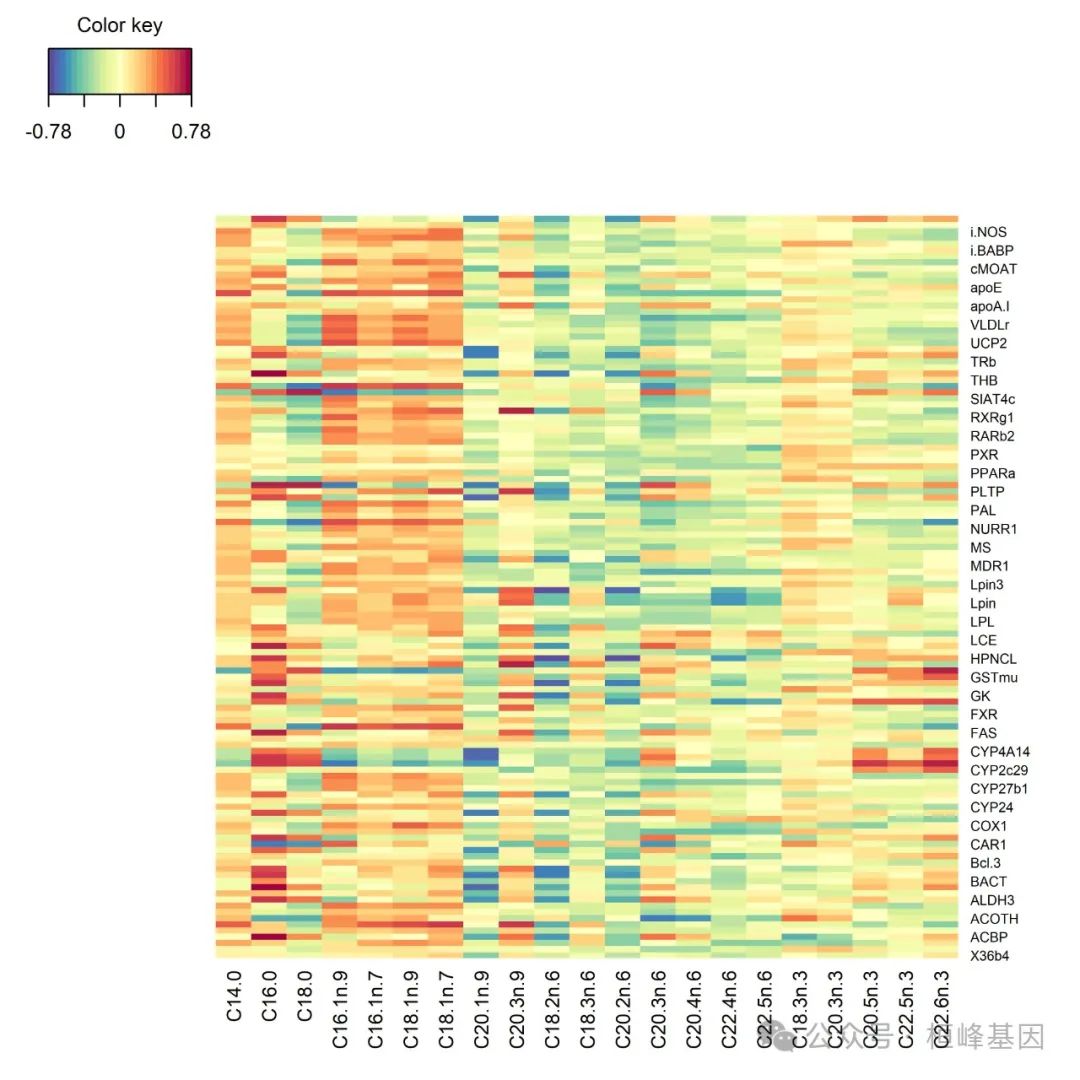
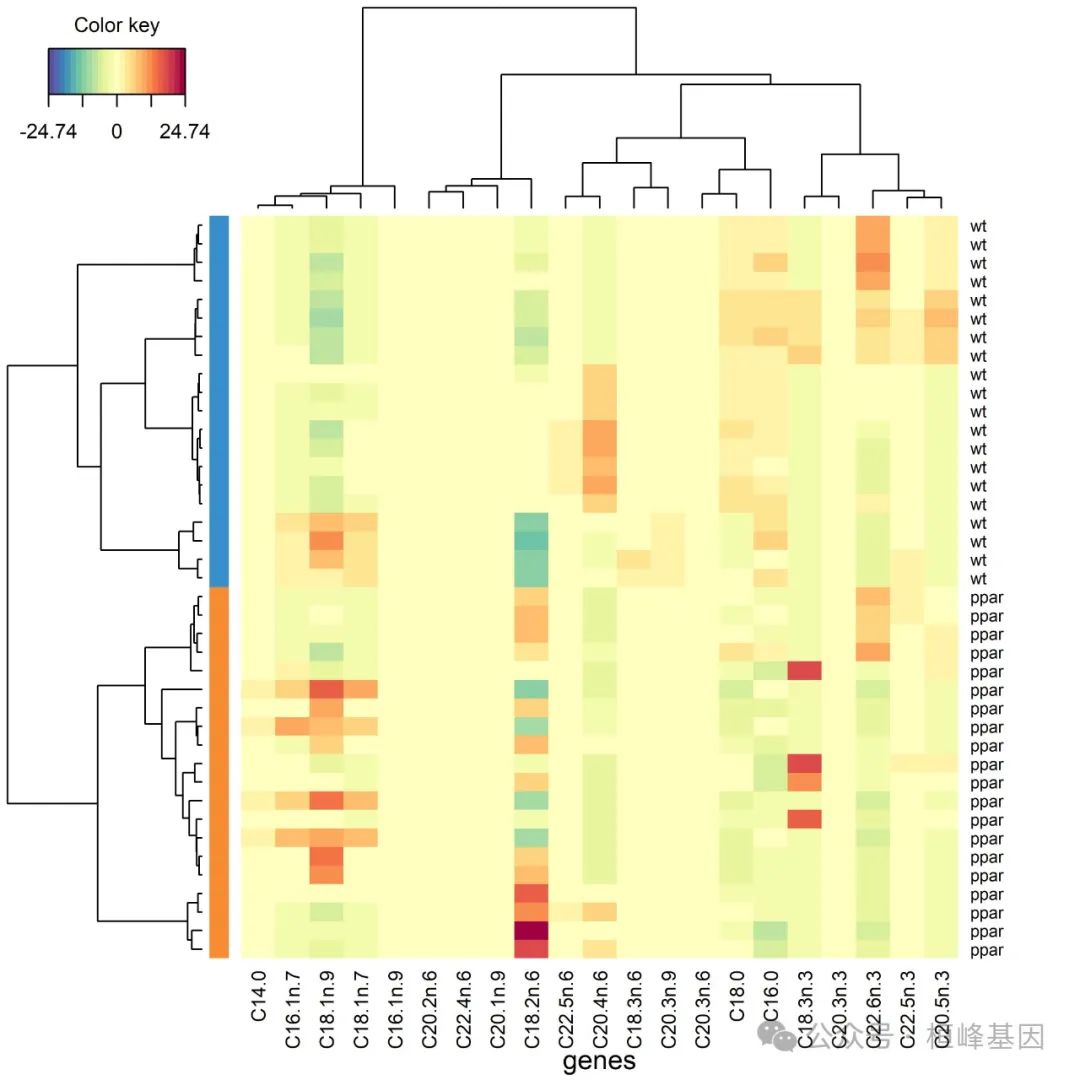
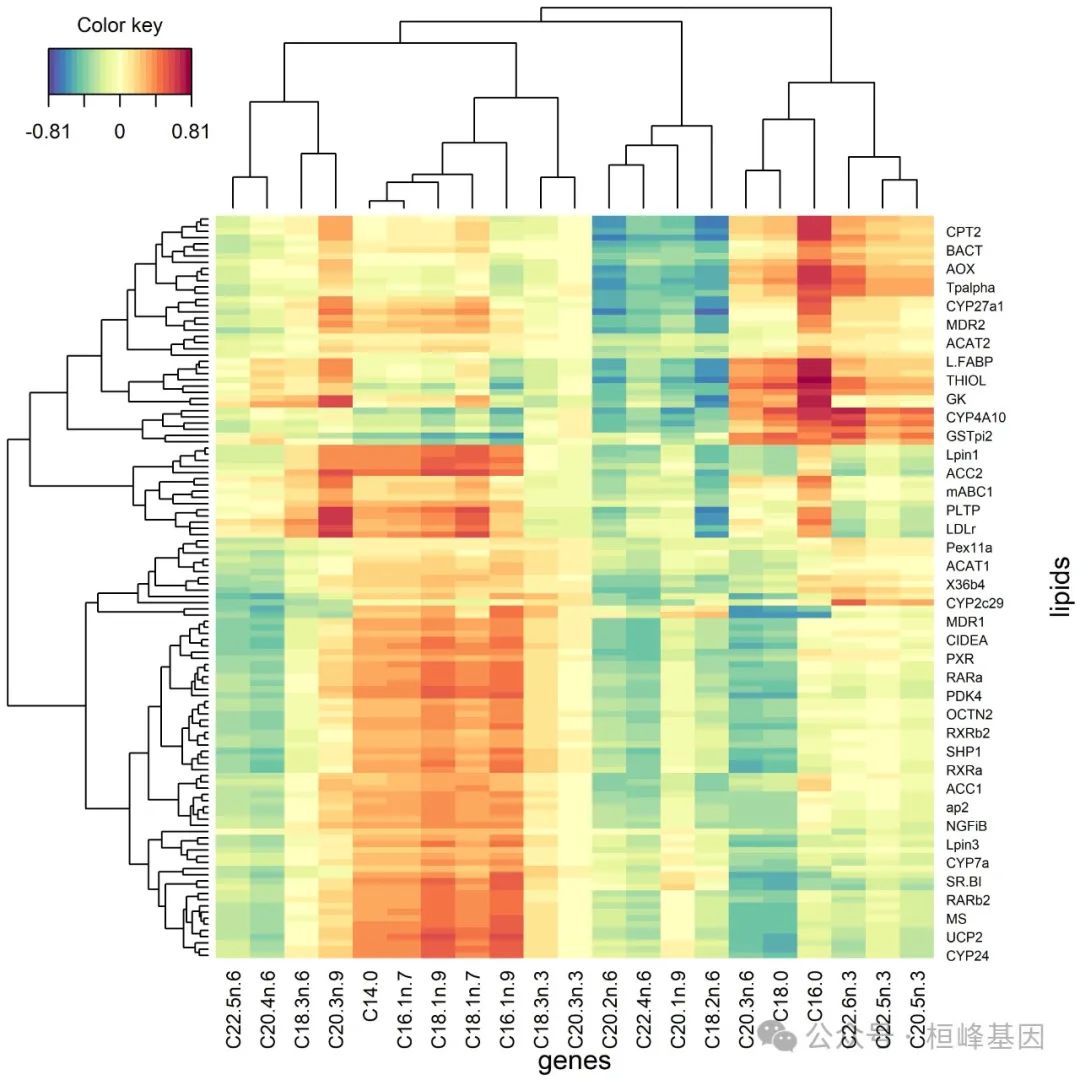
结果导出
# 保存关键变量权重
write.csv(final_model$loadings$X, "gene_loadings.csv")
write.csv(final_model$loadings$Y, "lipid_loadings.csv")扩展说明
关键参数说明
参数 | 功能描述 |
|---|---|
ncomp | 设置分析的主成分数量 |
keepX/Y | 控制每个成分保留的变量数 |
mode | 指定分析模式 (regression/canonical) |
cor | 计算变量间相关系数阈值 |
使用建议
数据预处理:
建议进行log转换和标准化
缺失值处理可使用
nipals方法
可视化优化:
使用
color.mixo()获取预设配色方案通过
cex参数调整图形元素大小
高级分析:
时间序列数据可使用
block.pls方法分类结局可用
block.splsda方法
Reference
Rohart F., Gautier, B, Singh, A and Lê Cao, K. A. mixOmics: an R package for ‘omics feature selection and multiple data integration. PLOS 2-17.

















 4757
4757

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









