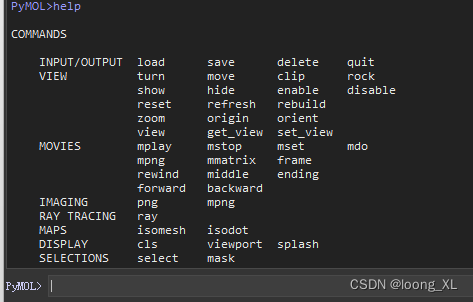
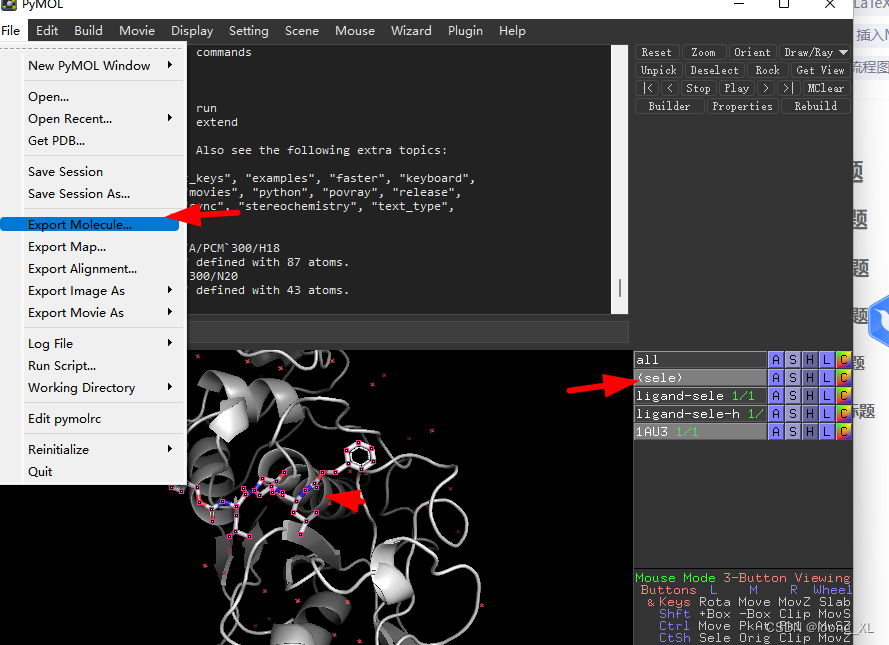
参考:https://www.jianshu.com/p/2b8e700ff37a 1、pymol 常见命令 可以输入help查看 help create remove solvent ##去除水分子 remove hydrogens ##去氢 remove organic ##分离得到蛋白 set_name old_name,new_name 2、分别保存小分子、蛋白 1)小分子保存 点选好小分子,选择保持小分子sele,格式可以选择pdb、mol2等








 超级会员免费看
超级会员免费看
 这篇博客介绍了如何在pymol中进行常见操作,包括保存小分子和蛋白质、分离蛋白多聚体亚基、创建对接位点的交互图以及去除杂原子。在保存小分子时要注意pymol加载pdb文件可能导致键信息错误,建议从rcsb网站单独下载小分子。此外,详细步骤展示了如何显示和标记氢键、选择和优化显示效果。
这篇博客介绍了如何在pymol中进行常见操作,包括保存小分子和蛋白质、分离蛋白多聚体亚基、创建对接位点的交互图以及去除杂原子。在保存小分子时要注意pymol加载pdb文件可能导致键信息错误,建议从rcsb网站单独下载小分子。此外,详细步骤展示了如何显示和标记氢键、选择和优化显示效果。


 订阅专栏 解锁全文
订阅专栏 解锁全文



















 1106
1106

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言


 被折叠的 条评论
为什么被折叠?
到【灌水乐园】发言
被折叠的 条评论
为什么被折叠?
到【灌水乐园】发言








