








RDKit|分子读取
Github: 地址
读SMILES/SMARTS
m = Chem.MolFromSmiles('C[C@H](O)c1ccccc1')
m = Chem.MolFromSmarts('Cc1ccccc1')
m
文件批量读取
从.csv批量读取:SmilesMolSupplier(data, delimiter, smilesColumn, nameColumn, titleLine, sanitize)
data:数据文件
delimiter:分隔符,默认为’ ’
smilesColumn:SMILES所在列,默认为0
nameColumn:SMILES名称所在列,默认为1
titleLine:是否含有标题行,默认True
sanitize:是否检查正确性,默认True
suppl = Chem.SmilesMolSupplier(data="./data/batch.csv", delimiter=",")
smiles = [Chem.MolToSmiles(m) for m in suppl]
print(smiles)
with open("./data/batch.csv", "r", encoding="utf-8") as f:
content = f.read()
suppl = Chem.SmilesMolSupplierFromText(text=content, delimiter=",")
smiles = [Chem.MolToSmiles(m) for m in suppl]
print(smiles)
DataFrame批量读取
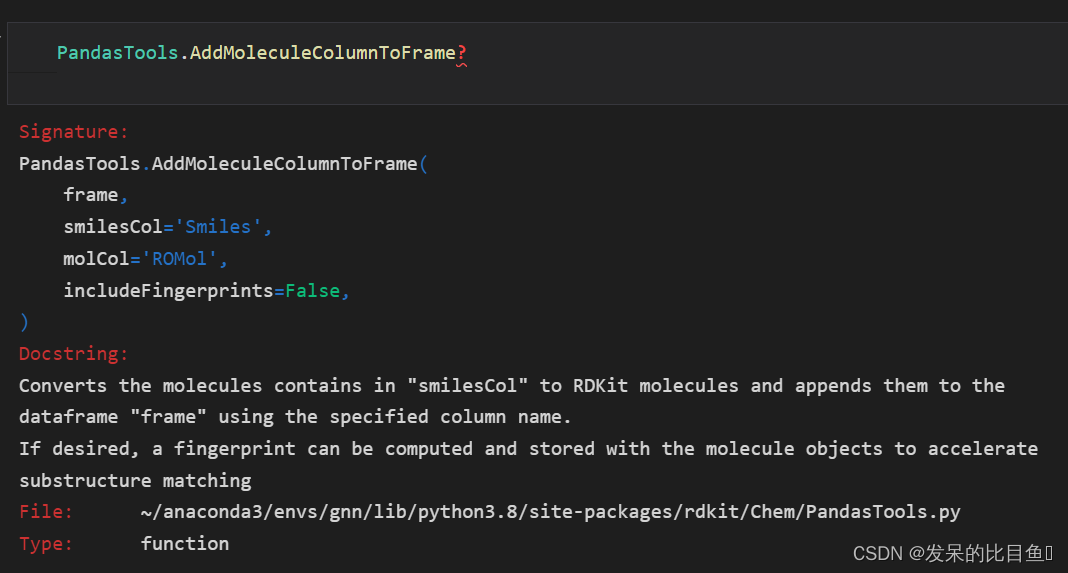
读取DataFrame中的SMILES:AddMoleculeColumnToFrame(frame, smilesCol, molCol, includeFingerprints)
frame:DataFrame对象
smilesCol:SMILES所在列
molCol:新列名,将存放产生的rdkit mol对象
includeFingerprints:是否生成指纹
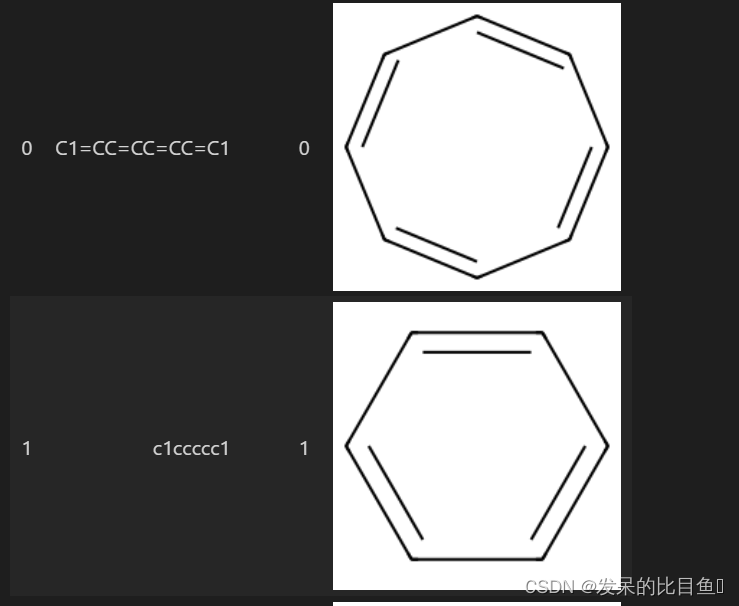
from rdkit.Chem import PandasTools
import pandas as pd
df = pd.read_csv('./data/batch.csv')
PandasTools.AddMoleculeColumnToFrame(frame=df,smilesCol='SMILES', molCol='mol' ,includeFingerprints=True)
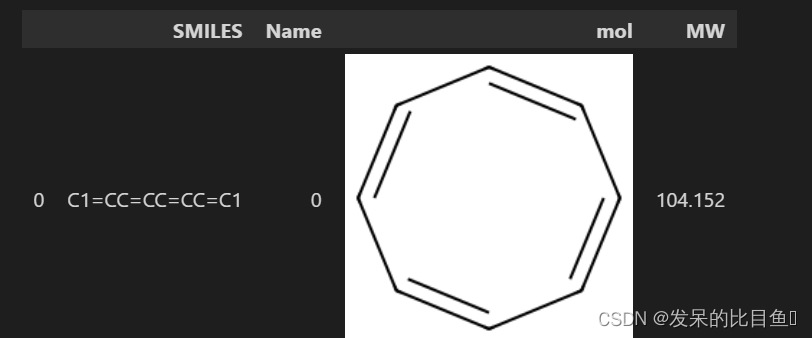
下面我们可以计算分子的质量
from rdkit.Chem import Descriptors
df["MW"] = df["mol"].apply(Descriptors.MolWt)
df
从.sdf里批量读取:SDMolSupplier(fileName, sanitize, removeHs, strictParsing)
fileName:文件名
sanitize:检查化合价,计算芳香性、共轭、杂化、kekule,默认True
removeHs:是否隐藏氢原子,默认True
strictParsing:是否使用严格模式进行解析,默认True
suppl = SDMolSupplier("./data/batch.sdf")
smiles = [Chem.MolToSmiles(m) for m in suppl]
print(smiles)
从压缩包file object/.gz里读取
import gzip
gz_file = gzip.open("./data/batch.sdf.gz", "r")
suppl = Chem.ForwardSDMolSupplier(gz_file)
smiles = [Chem.MolToSmiles(m) for m in suppl]
print(smiles)
读.mol
从.mol里读取:MolFromMolFile(fileName, sanitize, removeHs, strictParsing)
m = Chem.MolFromMolFile('./data/single.mol')
m
读.mol2
不推荐,容易出bug:MolFromMol2File(…)
m = Chem.MolFromMol2File('data/batch.mol2')
print(Chem.MolToSmiles(m))
读取pdb
mol = Chem.MolFromPDBFile("./data/single.pdb")
print(Chem.MolToSmiles(mol))
mol =Chem.MolFromPDBBlock("""COMPND UNNAMED
AUTHOR GENERATED BY OPEN BABEL 3.1.1
HETATM 1 C UNL 1 0.000 0.000 0.000 1.00 0.00 C
HETATM 2 C UNL 1 0.000 0.000 0.000 1.00 0.00 C
HETATM 3 C UNL 1 0.000 0.000 0.000 1.00 0.00 C
HETATM 4 C UNL 1 0.000 0.000 0.000 1.00 0.00 C
HETATM 5 C UNL 1 0.000 0.000 0.000 1.00 0.00 C
HETATM 6 C UNL 1 0.000 0.000 0.000 1.00 0.00 C
HETATM 7 C UNL 1 0.000 0.000 0.000 1.00 0.00 C
HETATM 8 C UNL 1 0.000 0.000 0.000 1.00 0.00 C
CONECT 1 8 2 2
CONECT 2 1 1 3
CONECT 3 2 4 4
CONECT 4 3 3 5
CONECT 5 4 6 6
CONECT 6 5 5 7
CONECT 7 6 8 8
CONECT 8 7 7 1
MASTER 0 0 0 0 0 0 0 0 8 0 8 0
END""")
print(Chem.MolToSmiles(mol))
读取fasta序列
mol = Chem.MolFromFASTA(""">3CA7_1|Chain A|Protein spitz|Drosophila melanogaster (7227)
TFPTYKCPETFDAWYCLNDAHCFAVKIADLPVYSCECAIGFMGQRCEYKEID""")
mol
mol = Chem.MolFromSequence("TFPTYKCPETFDAWYCLNDAHCFAVKIADLPVYSCECAIGFMGQRCEYKEID")
mol
读取Inchi
mol = Chem.MolFromInchi("InChI=1S/C8H10O/c1-7(9)8-5-3-2-4-6-8/h2-7,9H,1H3/t7-/m0/s1")
print(Chem.MolToSmiles(mol))



















 4645
4645

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











