







 GWAS分析中,增加样本量能提升检验效能,尤其对检测低频和罕见变异位点至关重要。当单个项目难以获取足够样本时,可通过meta分析合并多个GWAS项目结果。本文介绍了meta分析的概念、常用软件以及模型选择,并探讨了其在基因组领域的应用。
GWAS分析中,增加样本量能提升检验效能,尤其对检测低频和罕见变异位点至关重要。当单个项目难以获取足够样本时,可通过meta分析合并多个GWAS项目结果。本文介绍了meta分析的概念、常用软件以及模型选择,并探讨了其在基因组领域的应用。
欢迎关注”生信修炼手册”!
对于GWAS分析而言,增加样本量是提高检验效能的最直接有效的方式。目前常规GWAS项目的样本量约为1000 cases vs 1000 controls,这样的样本量能够检测到的相关SNP位点基本属于common SNP, 频率在1%以上,对应的OR值也通常在1.2以上,对于低频和罕见突变位点,常规的样本量则无法有效检出,因为携带对应Allel的样本太少,很难达到统计学显著性。
对于多基因复杂疾病而言,其相关联的的SNP位点肯定不仅限于common SNP, 为了有效检测相关联的低频和罕见变异位点,需要进一步增加GWAS分析的样本量,然而考虑到样本收集的难度,周期和实验成本,单个项目很难达到有效的样本量。鉴于这个情况,最直接的解决方案就是合并多个GWAS项目的结果,来达到增加样本量的目的。多个数据集的合并分析,正是meta分析大展身手的时候。
meta-analysis, 称之为元分析,或者荟萃分析,早在1976年就提出了这个概念,其分析对象是已有的研究成果,用来对先前研究进行综合评价和定量合并,在多个领域都有其应用。在生命科学领域,包括基因组,转录组,蛋白质组等多组组学研究i领域,都有meta分析的用武之地,而GWAS的meta 分析就是meta分析在基因组领域的最经典应用场景。
meta分析有多种算法,比如基于pvalue, 基于排序,基于效果量等各种算法,每种算法各有优劣。时至今日,相关的工具和软件也很多,对于GWAS的meta分析而言,METAL, PLINK, MetaSoft等软件都可以实现对应的功能,
在整合多个数据集的分析结果时,我们需要考虑到不同数据集之间的差异,也称之为数据集的异质性,常用的有以下两种模型
Fixed effects models, FEM
Random effects models, REM
第一种称之为固定效应模型,第二种称之为随机效应模型,固定效应模型中假设不同数据集的差异是固定,而随机效应模型假设不同数据集的差异服从正态分布,因此,固定效应模型适用于相同实验条件下的数据,而随机效应模型则可以用于处理不同来源的独立数据。
对于GWAS meta分析的结果,经典的可视化方式如下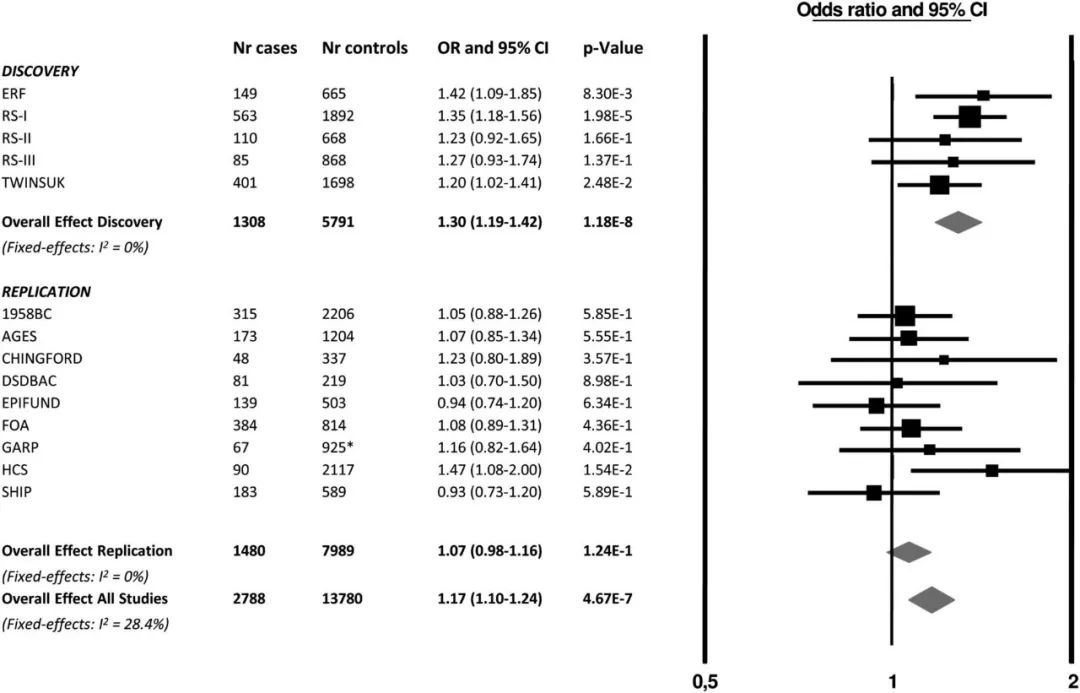
这种图称之为forest plot, 很好了展示了每个study中变异位点对应的OR和P值,在后面的文章中,会详细介绍各个软件进行meta分析的用法,敬请期待!
·end·
—如果喜欢,快分享给你的朋友们吧—
往期精彩
基因型填充
CNV分析
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 3199
3199

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









