






网站:http://www.sxdyc.com/diffDeseq2Analyse
一、deseq2差异分析简介
DESeq2是一个为高维计量数据的归一化、可视化和差异表达分析而设计的一个R语言包。它通过经验贝叶斯方法(empirical Bayes techniques)来估计对数倍数变化(log2foldchange)和离差的先验值,并计算这些统计量的后验值。
二、使用须知(几个概念)
1、Group:表示的是样本的分组信息。差异分析中,一般只有两组,进行比较,即A和B组进行比较;
2、P值:P值即概率,反映某一事件发生的可能性大小。在差异分析中,p值的大小反应的是分组样本的重复性,组内重复性越好,p值往往越小,在分析的过程中,我们一般认为满足p<0.05的情况下,该特征(基因)差异才是真正的差异基因,而不是由于离群样本过高/过低导致的假阳性结果。
三、使用方法
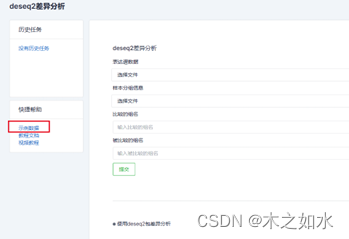
1、打开网址(http://www.sxdyc.com/singleCollectionTool?href-diff),选择“deseq2差异分析”。
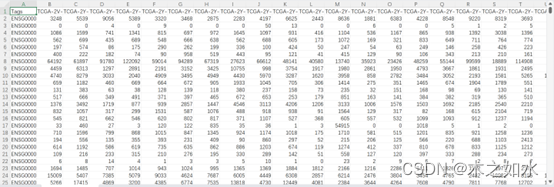
2、准备数据
一个全基因的表达谱矩阵,其中行为基因,列为样本
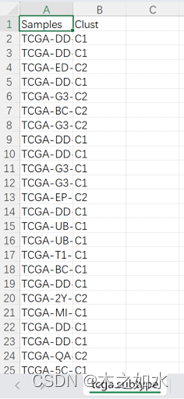
一个样本分组信息,包含两列,第一列为样本名,第二列的分组
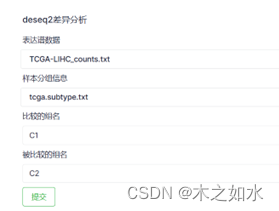
3.输入比较和被比较的组名
这里输入的是比较组为C1,被比较组为C2,代表该差异分析为C1vsC2
获取到的结果,log2(FC)>0的基因为在C1组中高表达,log2(FC)<0的基因为在C1组中低表达
这里需要注意的是deseq2包进行差异分析输入的readcount的数据,而不能是TPM/FPKM的数据。
数据格式用为txt文本,以制表符分割。
4.点击提交
5.输入分析队列名,点击提交
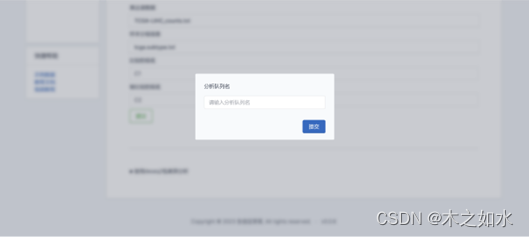
- 等待结果,查看结果
结果生成了两个文件:
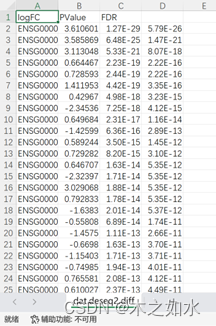
dat.deseq2.diff.txt (差异分析的结果)
结果需要注意的是:列名错位,A列其实是基因,B为logFC,C为PValue,D为FDR
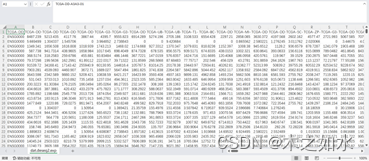
dat.deseq2.exp.txt (预处理后的基因的表达谱)
四、分析结果
- log2FC中的FC即 fold change,表示两样品(组)间表达量的比值,对其取以2为底的对数之后即为log2FC。
- FDR即False Discovery Rate,错误发现率,是通过对差异显著性p值(p-value)进行校正得到的。
注意:在自测数据中,由于样本较少,在选择差异分析时,可以选择p值而不是FDR(校正后的p值)
当然,如果不清楚数据是什么样的,可以选择下载我们的示例数据,可以关注公众号:豆芽数据分析















 3万+
3万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









