



ChIPQC 是 Bioconductor 旗下用于评估 ChIP-seq 数据质量的 R 包,可自动化计算关键指标并生成可视化报告。它通过分析测序 reads 在基因组上的分布特征,量化富集效果、背景噪声及实验重复性,为后续分析提供质量基准。
但是其基因注释信息是基于老版本基因组的比如 mm10 和 hg19,为了和 ChIP-seq 上有分析使用的基因组版本一致,这里自行构建基因注释信息用于报告中的绘图展示,如果没有这个注释信息,会有几张图是空白的,报告也不让太完整,但是影响也不大,比较我们可以使用其他工具比如 ChIPSeeker 注释 Peak 的位置注释。
安装方法
if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager")
BiocManager::install("ChIPQC")
BiocManager::install("TxDb.Mmusculus.UCSC.mm39.knownGene")
BiocManager::install("TxDb.Hsapiens.UCSC.hg38.knownGene")
输入准备
需准备包含样本元数据的 TSV 文件,一般包含以下字段:
SampleID:样本唯一标识符 ControlID:与SampleID 样本对应的 Input 样本的标识符 Factor:分组信息,比如实验组与对照组 bamReads: BAM 文件路径 bamControl:Input 样本的BAM 文件路径 PeakFile:峰值文件路径(可选,支持 BED、narrowPeak 等格式) PeakCaller:"narrow":macs 的*_peaks.narrowPeak; "bed":macs 的*_peaks.xls 文件,即宽峰和窄峰不一样 Replicate:生物重复样本编号
experiment.tsv 文件如下所示,我们需要再此基础上增加其他列
SampleID Factor ControlID Replicate
KO_1 KO KO_1.input 1
KO_2 KO KO_2.input 2
WT_1 WT WT_1.input 1
WT_2 WT WT_2.input 2
增加bamReads、bamControl、PeakCaller、Peaks 列信息
peak_format = "narrow"
meta <- read.table('experiment.tsv',header = T)
meta$bamReads = paste0("Result/02.Alignment/",meta$SampleID,".bam")
meta$bamControl = paste0("Result/02.Alignment/",meta$ControlID,".bam")
if(peak_format=="narrow"){
meta$PeakCaller = "narrow"
meta$Peaks = glue::glue("Result/03.Peak_Calling/{peak_format}Peak/{meta$SampleID}_peaks.narrowPeak")
}else if(peak_format=="broad"){
meta$PeakCaller = "macs"
meta$Peaks = glue::glue("Result/03.Peak_Calling/{peak_format}Peak/{meta$SampleID}_peaks.xls")
}
自定义 mm39 基因注释信息
annotation 便是mm39 的基因注释信息
specie = "mmu"
txdb=switch(specie,
mmu=TxDb.Mmusculus.UCSC.mm39.knownGene::TxDb.Mmusculus.UCSC.mm39.knownGene,
hsa=TxDb.Hsapiens.UCSC.hg38.knownGene::TxDb.Hsapiens.UCSC.hg38.knownGene
)
All5utrs <- reduce(unique(unlist(fiveUTRsByTranscript(txdb))))
All3utrs <- reduce(unique(unlist(threeUTRsByTranscript(txdb))))
Allcds <- reduce(unique(unlist(cdsBy(txdb,"tx"))))
Allintrons <- reduce(unique(unlist(intronsByTranscript(txdb))))
Alltranscripts <- reduce(unique(transcripts(txdb)))
posAllTranscripts <- Alltranscripts[strand(Alltranscripts) == "+"]
posAllTranscripts <- posAllTranscripts[!(start(posAllTranscripts)-20000 < 0)]
negAllTranscripts <- Alltranscripts[strand(Alltranscripts) == "-"]
chrLimits <- seqlengths(negAllTranscripts)[as.character(seqnames(negAllTranscripts))]
negAllTranscripts <- negAllTranscripts[!(end(negAllTranscripts)+20000 > chrLimits)]
Alltranscripts <- c(posAllTranscripts,negAllTranscripts)
Promoters500 <- reduce(flank(Alltranscripts,500))
Promoters2000to500 <- reduce(flank(Promoters500,1500))
LongPromoter20000to2000 <- reduce(flank(Promoters2000to500,18000))
annotation=list(
version="mm39",
LongPromoter20000to2000=LongPromoter20000to2000,
Promoters2000to500=Promoters2000to500,
Promoters500=Promoters500,
All5utrs=All5utrs,
Alltranscripts=Alltranscripts,
Allcds=Allcds,
Allintrons=Allintrons,
All3utrs=All3utrs
)
生成报告
一行代码生成报告
ChIPQC::ChIPQC(meta, annotation=annotation) %>%
ChIPQCreport(chipObj, reportName="ChIP QC report", facetBy="Factor",reportFolder="ChIPQCreport")
报告解读
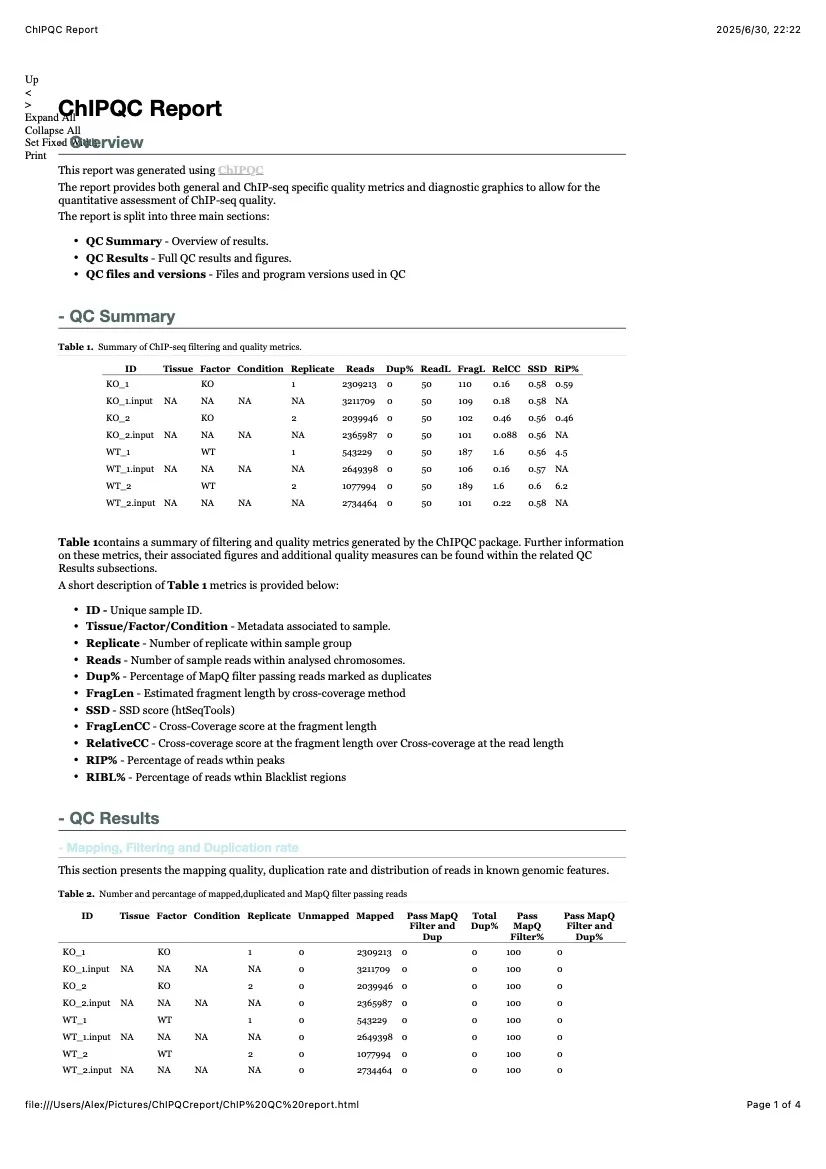
生成的 HTML 报告包含多个关键模块:
比对统计:展示测序深度、映射率及染色体分布 富集评估:通过 FRiP 分数(Peaks 区域 reads 比例)评估抗体特异性 峰质量:分析峰宽分布、峰强度及峰重叠率 可重复性:基于 IDR 值评估生物学重复间的一致性 基因组分布:显示 reads 在启动子、外显子等功能区域的比例 批次效应:PCA 分析检测样本间潜在的技术变异
关键指标阈值参考:
RIP% > 0.1:提示有效富集 SSD < 0.8:表明信号分布集中
低质量指标可能暗示实验操作问题(如抗体效价不足)或测序深度不够,需结合具体指标进行实验优化。




















 3324
3324

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









