










Comparative transcriptomic analysis unveils the deep phylogeny and secondary metabolite evolution of 116 Camellia plants ,比较转录组分析的一篇文献。
SUMMARY
茶花植物包括200多个种类,具有极大的多样性和巨大的经济、观赏和文化价值。我们对几乎所有茶属(Camellia)的分支的116种茶花植物进行了转录组测序。我们构建了一个含有89,394个基因家族的茶花植物泛转录组,然后基于405个高质量的低拷贝核心基因解决了茶属的系统发育关系。大多数推断的关系通过多个核基因树和形态特征得到了很好的支持。我们提供了强有力的证据表明,茶花植物共享了一个近期的全基因组复制事件,随后是与抗压力和次生代谢相关的转录因子家族的大规模扩张。特别是与茶叶品质相关的次生代谢产物,如儿茶素和咖啡因,在来自茶节(Thea)的茶花植物中被优先大量积累。我们彻底检查了与茶叶品质相关的数百个基因的表达模式,并发现其中一些在Thea种中表现出显著高的表达量和与次生代谢物积累的相关性。我们还发布了一个可通过网络访问的数据库,以便高效检索茶花转录组。报告的转录组序列和获得的新发现将促进茶花种质资源的有效保护和利用,支持培育茶树、山茶和油茶植物的育种项目。
INTRODUCTION
茶属是山茶科中最大的属,根据张氏(Chang, 1981)分类系统,包含18个分部的200多个种类。这些种类具有重要的经济和观赏价值,广泛用于生产茶叶、油料和观赏花卉(夏等,2017a)。例如,属于茶属Thea分部的Camellia sinensis(茶树)的叶子是茶叶的主要原料,茶是全球三大最受欢迎的非酒精饮料之一。属于Camellia分部的Camellia japonica(日本山茶)和Camellia reticulate(网纹山茶)的花朵是重要的盆栽和园林产品,在全球花卉贸易中扮演着必不可少的角色。此外,Camellia oleifera(油茶)是中国重要的木本油料植物,属于茶属Oleifera分部。其果实通常用于生产食用油,具有巨大的经济和健康价值。如何有效加速茶花植物的利用和保护,以促进茶叶、山茶和茶油产业的发展,代表着一个最紧迫但仍是长期未解决的科学问题,主要是由于茶花植物的系统发育关系不明确和对影响茶花植物品质相关的次生代谢物演化机制理解不足。
目前,有三个著名的茶花植物分类系统,分别由Sealy(1958)、Chang(1981)和Ming(1999)提出。它们的分类标准主要基于茶花植物的形态学、孢粉学、染色体和生物地理学证据,但关于亚属、分部和种的划分仍存在许多争议。近年来,随着进化生物学的发展和创新,与传统的古典分类方法(江等,2010;卢等,2012;皮等,2009)相比,分子生物学的研究方法已逐渐引入茶花植物的系统发育研究,拓宽了我们对这个属的分类、进化和系统发育的了解。到目前为止,关于茶花植物系统发育的大多数研究主要使用叶绿体(如matK、rbcL、trnL-F、rpL16和psbA-trnH)或线粒体(如matR)DNA片段,或整个器官基因组序列(陈等,2014;杨等,2005;杨等,2006;杨等,2013)。然而,由于茶花植物器官的基因组序列相对保守,进化速率缓慢(黄等,2014),加之样本收集的代表性不足和使用的基因片段数量少,为系统发育重建发现的信息位点较少,导致器官基因序列在茶花植物系统发育调查中的应用受限。此外,在植物系统发育研究中常用的分子标记,如核糖体DNA的转录间隔区(ITS)(唐等,2004;Vijayan & Tsou,2008)、低拷贝核基因(如RPB2、waxy和PAL)(刘等,2012;肖和Parks,2003;杨等,2006
)和随机扩增多态性DNA标记(陈和山口,2002;史等,1998),也广泛用于茶花植物的系统发育研究。然而,这些研究得到的系统发育树支持度较差,拓扑结构需要进一步加强,这不足以获得对茶花植物系统发育和进化的全面理解。
与叶绿体和线粒体基因片段相比,通过基因组和/或转录组测序获得的大量核基因序列提供了丰富的低拷贝核基因和信息位点,用于构建系统发育树,并已广泛用于解决许多重要和复杂植物群体的深层系统发育,如被子植物(曾等,2014)、葡萄科(文等,2013)、兰科(邓等,2015)、十字花科(黄等,2016)、石竹目(杨等,2015)、唇形目(张等,2020a)和真双子叶植物(曾等,2017)。此外,茶植物富含次生代谢物,如儿茶素、茶氨酸和咖啡因。它们不仅赋予茶叶色泽、香气和口感的品质特征,还对人类健康有益(夏等,2019;张等,2019)。然而,这些与品质特征相关的次生代谢物如何在茶花植物的进化过程中积累和演化仍然不清楚。在一个解决良好的系统发育框架下揭示茶花植物次生代谢物的演化模式将有助于高效保护和利用这些珍贵的茶花种质资源,促进茶叶、山茶和茶油产业的发展。
在本研究中,我们收集并使用Illumina测序技术(Illumina Inc.,圣地亚哥,加州,美国)测序了几乎所有茶属分部的116种茶花植物的转录组。我们还通过高效液相色谱(HPLC)测定了10多个类别的次生代谢物的含量。通过对测序数据的深入分析,结合比较转录组学、系统发育和植物化学研究,我们旨在获得关于茶花植物复杂的系统发育、基因组演化和次生代谢物多样化的新见解。
RESULTS
Transcriptome sequencing, assembly and characterization
我们对116株山茶属(Camellia)植物的转录组进行了测序,这些植物代表了山茶属94%的分类组,使用的是Illumina Novaseq平台。我们还测序了Polyspora speciosa(山茶属的外群)的转录组。此外,我们从NCBI数据库(National Center for Biotechnology Information)下载了九个栽培茶树(C. sinensis)品种的测序数据集(表S1)。总共生成了3.48亿对端清洁读取和899.25 Gb的清洁基底,平均每个物种有7.69 Gb(表S1)。使用trinity (Haas et al., 2013) 进行de novo转录组组装,共生成了15,998,227个转录本,平均长度和N50大小分别为695 bp和1089 bp(图1a,b, 表S2)。我们使用busco(Benchmarking Universal Single Copy Orthologs)(Simão et al., 2015)评估了转录组组装的完整性。我们发现,平均而言,94.56%的植物保守直系同源基因出现在组装的转录组中,显示了转录组组装的良好质量(图1c和表S3)。
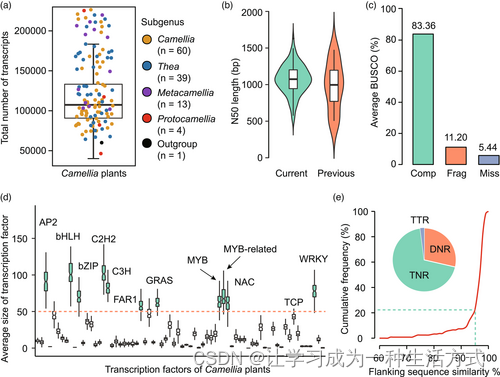
116株山茶属植物转录组的特征。(a) 每株山茶属植物组装的转录本总数。(b) 当前组装与先前报告之间N50大小的比较。(c) 使用BUSCO评估转录组组装的完整性。展示了三种类型的BUSCO的比例:完整(Comp),碎片(Frag),和缺失(Miss)。(d) 不同山茶属植物中转录因子(TFs)的分布。红色虚线代表平均大小大于50的转录因子家族,在箱线图中以绿色填充。(e) 山茶属植物多态性SSR的侧翼区域序列相似性。饼图表示多态性SSR类型的比例,包括二核苷酸重复(DNRs),三核苷酸重复(TNRs),和四核苷酸重复(TTRs)。
接着,我们使用六个公共蛋白数据库为组装的山茶属转录本注释了推定功能,包括Swissprot (www.uniprot.org);PFAM (Pfam is now hosted by InterPro);京都基因与基因组百科全书(KEGG)(www.genome.jp/kegg);基因本体论(GO)(Gene Ontology Resource);COG (www.ncbi.nlm.nih.gov/COG);以及TAIR10 (www.arabidopsis.org)。结果显示,89.65%(14,297,114)的总转录本可以用已知的蛋白质和/或域进行注释(表S4)。转录因子(TFs)的注释产生了196,809个含TF的转录本,平均每个山茶属物种有1682个(表S5)。MYB/MYB相关(17,002)是山茶属转录组中最重要的TF家族,其次是C2H2(13,331)和bHLH(13,306)(图1d)。我们还在山茶属转录组中鉴定出5,322,939个简单序列重复(SSR)(图1e和表S6)。其中,有786个SSR在山茶属植物中是多态性的,为茶树的未来基因组学和育种研究提供了宝贵的遗传资源(表S7)。
Comparative transcriptomic analysis resolves the phylogeny and origin of genus Camellia
山茶属植物的高质量和全面采样的转录组使我们能够彻底研究山茶属植物的全转录组(总转录本集合)和核心转录组(所有物种共享的转录本)的特性。随着山茶属转录组数据集的增加,我们观察到总转录本数量增加,但共享转录本数量减少(图S1a)。任意两个山茶属分类组的全转录组大小平均解释了所有116种山茶属物种内发现的32.64%,表明单一的转录组和/或基因组无法充分代表整个山茶属的遗传多样性。全转录组的大小主要取决于山茶属植物的总数,最初的山茶属全转录组构建包含89,394个正交群,代表了116种山茶属植物的遗传多样性(图S1b)。我们还发现,山茶属全转录组中大约有5793个正交群是核心群组,这些核心群组在所有116种山茶属植物中共享,而77,874个正交群是非核心群组,这些非核心群组出现在超过一种但不是所有116种山茶属转录组中。正如预期的那样,仅在一个样本中发现的独特正交群组频率最低,而在所有116种山茶属植物中,C. cordifolia具有最多的物种特异性基因组(表S8)。此外,我们展示了核心山茶属转录本(99.01%)比非核心转录本(92.40%)在NCBI NR数据库中更有可能注释出更多同源序列(图S1c)。
构建的全转录组使我们能够真正解决山茶属的系统发育和分化问题——这是多年来困扰茶叶社区的一个问题(Xia et al., 2020c)。我们从核心山茶属OGs中鉴定出总共405个低拷贝同源基因(OGs),然后进行多重比对以构建山茶属的系统发育(图S1d)。使用raxml(Stamatakis, 2014)和mrbayes(Ronquist et al., 2012)进行的系统发育分析产生了几乎相同的拓扑结构,并得到了强有力的支持,显示山茶属的系统发育可以被划分为七个簇(图2)。大多数分类组,如Thea和Furfuracea,被恢复为单系群,支持率为100%。与明的分类系统一致,Glaberrima和Thea部分之间的物种,以及Paracamellia和Oleifera部分之间的物种显示了密切的关系,支持了它们的分类组合。值得注意的是,我们发现Pseudocamellia和Tuberculata部分的物种聚集成一个单一的支系,而Eriandria部分的Camellia lawii与Theopsis部分的物种关系最为密切。此外,Chrysantha部分位于构建的系统发育的基部,展示了山茶属最古老的部分。不同于明和张的分类系统,我们展示了Luteoflora部分的C. luteoflora与Tuberculata部分的物种接近,而不是与Camellia部分的物种聚集的Stereocarpus部分。此外,获得的系统发育树还支持了《中国植物志》对许多代表性山茶属物种的修订,如Camellia kwangtungensis、Camellia danzaiensis和Camellia chungkingensis,这反过来暗示了系统发育树的准确性和稳健性(Ming & Bartholomew, 2007)。
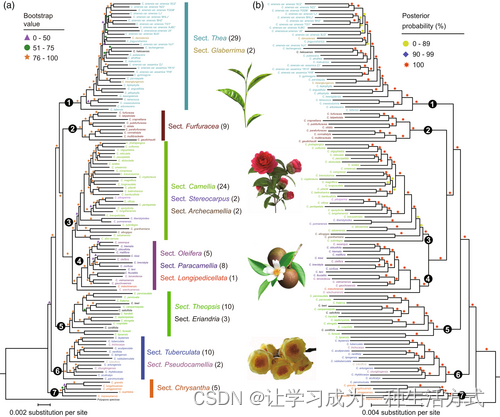
使用最大似然法和贝叶斯方法构建的116种山茶属植物的系统发育。(a)最大似然树。(b)贝叶斯树。引导值和后验概率以颜色标记并显示在系统发育树的分支上。山茶属的主要分类组在树旁标记。括号中的数字表示本研究中使用的物种数量。
清晰解析的拓扑结构和深层关系为估计山茶属植物的分化时间提供了一个稳健的框架。我们估计山茶属植物起源于1400万年前(百万年前)(图3a)。Thea分类组植物的多样化可能起源于667万年前,即山茶属植物起源后763万年。有趣的是,栽培茶树(C. sinensis)两个变种的分化估计发生在82万年到216万年前,与使用一小部分共线基因估计的38万年到154万年前(Wei et al., 2018)一致,但晚于使用全基因组核基因估计的439万年前(Xia et al., 2020a)。我们还发现,Camellia和Oleifera分类组的多样化可以追溯到588万年前,那时它们从与Furfuracea分类组的共同祖先分离出来,大约在658万年前。
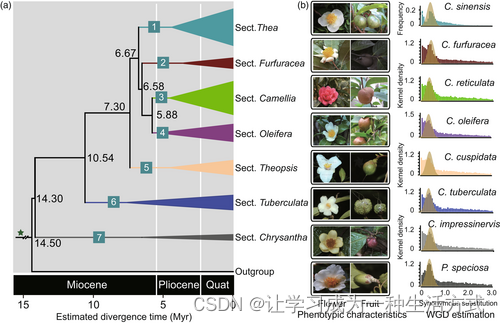
山茶属植物间分化时间及全基因组复制事件的估计。(a)分化时间估计。第四纪被简称。绿色星号表示全基因组复制(WGD)事件。(b)左侧面板显示了七种代表性山茶属植物及一个外群Polyspora speciosa的表型特征(花和果实),而右侧面板基于同源基因组Ks分布推断它们的WGD事件。注意,C. sinensis的WGD事件是基于基因组共线性分析推断的。
Recent whole genome duplication of camellia plant and its effects on the evolution of genes associated with quality traits
先前的研究发现,栽培茶树(C. sinensis)经历了近期的全基因组复制(WGD)事件(Wang et al., 2021; Xia et al., 2017b; Xia et al., 2020b),但这个WGD事件是否发生在其他山茶属植物中,以及它如何影响山茶属植物的品质特性仍然不清楚。对山茶属植物转录组的测序使我们能够彻底理解它们的WGD事件及其对表型多样性的影响。为了检查山茶属WGDs的进化历史,我们从系统发育树的代表性分支中选择了七种代表性的山茶属植物和一个外群物种P. speciosa,使用wgd软件包(Zwaenepoel & Van de Peer, 2019)鉴定它们的假定同源基因对。结果显示,平均每个物种获得了5120对同源基因对,占总基因的32.32%(表S9)。这些基因对的同义替代率(Ks)分布显示,所有七种代表性山茶属植物和外群物种都经历了与栽培茶树相同的WGD事件,Ks在0.36左右达到峰值(图3b),根据每位点每年6.5 × 10^−9的通用同义替代率(Gaut et al., 1996)计算,这大约是2800万年前,表明WGD事件发生在山茶属植物多样化之前(约1430万年前)。进一步分析猕猴桃的基因组序列发现,这次近期的WGD事件不是山茶属特有的,而是与其他杜鹃花目植物如猕猴桃(Ad-β;图S2)共享的。众所周知,茶树(C. sinensis)、茶油树(C. oleifera)和大红花山茶(Camellia reticulata)是山茶属三种最具经济价值的代表性作物,它们在茶叶和油以及观赏花卉的生产中具有关键应用(Xia et al., 2017a)。为了进一步了解WGD事件对与山茶属植物生理和表型特性相关的基因进化的影响,我们对三种代表性山茶属植物的WGD衍生的重复基因进行了功能注释,包括C. sinensis、C. oleifera和C. reticulata。结果显示,这些基因中的一些在与脂肪酸代谢过程(GO:0006631; P = 2.35 × 10^−11)、UDP-糖基转移酶活性(GO:0008194; P = 5.27 × 10^−11)、脂质生物合成过程(GO:0008610; P = 1.01 × 10^−10)、芳香化合物生物合成过程(GO:0019438; P = 1.66 × 10^−10)和色素代谢过程(GO:0042440; P = 3.01 × 10^−4)相关的GO类别中富集,表明山茶属植物最近的WGD事件是推动形成农艺性状和山茶属植物表型特性的关键动力(表S10)。
Well-resolved phylogeny of genus Camellia unveils the mechanisms underlying preferentially accumulation of tea quality related secondary metabolites in Camellia species from section Thea
植物次生代谢产物是常见于植物中的小分子化合物,不仅具有重要的经济和药用价值,而且作为植物系统发育深入理解的重要化学物质证据(张等,2020年)。山茶科植物富含次生代谢产物,被认为是次生代谢研究的重要植物群体(夏等,2020年)。儿茶素、茶氨酸和咖啡因是栽培茶树中三种重要且丰富的次生代谢产物,不仅赋予茶叶色、香、味等品质特征,而且有益于人体健康(张等,2019年)。茶属植物系统发育的良好解析使我们能够深入研究决定茶叶品质的次生代谢产物的分布和演化,并进一步探索茶属植物次生代谢产物的起源、演化和适应机制(图4a)。我们发现,与茶叶品质相关的次生代谢产物,尤其是儿茶素和咖啡因的含量在山茶科植物中变化较大,并且在茶属植物中倾向于在茶亚属物种中高度积累(图4b)。儿茶素是茶树的主要功能成分,属于多酚类,与茶味的苦涩和涩味有关。茶树中的儿茶素主要包括儿茶素(C)、(−)-表儿茶素(EC)、(−)-没食子儿茶素(GC)、(−)-表儿茶素-3-没食子酸酯(ECG)、(−)-表没食子儿茶素(EGC)和(−)-表没食子儿茶素-3-没食子酸酯(EGCG)。我们发现,尽管儿茶素在山茶科植物中普遍存在,但不同茶属植物组的物种中主导的儿茶素完全不同(表S11)。在茶亚属植物中,EGCG是最丰富的儿茶素,而在非茶亚属植物(如油茶)中,EC和EGC是最具代表性的儿茶素。有趣的是,茶亚属植物中的EGCG含量随着它们的分化而增加,但非茶亚属植物中没有这种趋势(图4b)。咖啡因是茶树中最常见的生物碱,与茶饮料的苦涩有关。与儿茶素类似,咖啡因在山茶科植物中普遍分布,尽管它们的含量有所不同。与非茶亚属植物相比,茶属植物中的茶亚属物种含有相当高的咖啡因含量,平均含量为14.65 mg/g(干重),范围从1.07 mg/g(C. tetracocca)到31.20 mg/g(C. angustifolia)。值得注意的是,分别是茶属系统发育基部和外类群的金花茶组和万灵花属植物,其咖啡因含量相对较低(平均为0.04 mg/g),与最近分化的茶亚属植物有最大差异(表S11)。茶氨酸是茶树中独特但非必需的氨基酸,它决定了绿茶的鲜味。我们证明了茶氨酸在一些非茶亚属的山茶科植物中未检测到,尽管在茶亚属植物中高度积累。在茶亚属植物中,基部植物似乎积累的茶氨酸比新分化的植物要低。此外,我们非常高兴地看到,次生代谢产物的进化模式与使用核基因重建的几个复杂茶属谱系的系统发育联系相辅相成(图4b)。例如,我们观察到茶属金花茶组和茶亚属的山茶科物种,以及疙瘩茶组和伪茶属之间的儿茶素、咖啡因和茶氨酸的含量分布类似,这进一步为这些组合处理提供了植物化学证据,如重建的系统发育所示。我们的结果还显示,在茶属植物中,三种特征代谢物的积累之间存在显著的正相关(表S12)。儿茶素和咖啡因在山茶科植物中的积累之间的皮尔逊相关系数(r)为0.91(P = 1.15 × 10^-33),而儿茶素和茶氨酸之间的相关系数为0.71(P = 5.43 × 10^-14)。
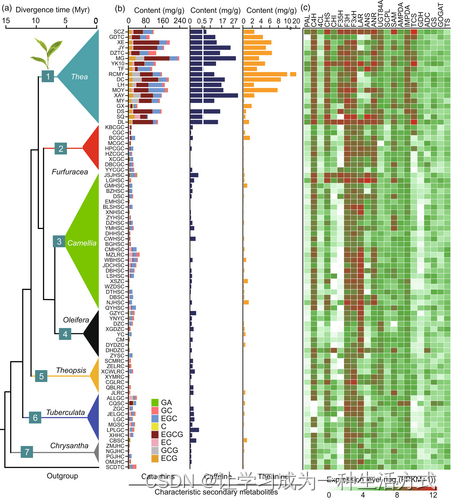
Camellia植物中与品质相关的次生代谢物质和基因表达模式的演化。(a)Camellia植物的系统发育。(b)不同Camellia植物中儿茶素(GC、EGC、C、EGCG、EC、GCG和ECG)、GA、咖啡因和茶氨酸含量(mg g^-1 干重)。(c)涉及儿茶素、咖啡因和茶氨酸生物合成的22个关键基因的表达模式。表达水平以log2(FPKM+1)评估,并使用R语言可视化。
为进一步探究山茶科植物次生代谢产物演化动态的分子机制,我们彻底研究了与儿茶素、咖啡因和茶氨酸生物合成相关的22个关键基因的表达模式,并将其与山茶科植物次生代谢产物的积累模式进行了相关性分析。结果显示,与非茶亚属的山茶科植物相比,大多数与品质相关的基因在茶亚属的物种中表达水平较高,这与山茶科植物次生代谢产物的积累模式一致(图4c)。特别地,我们发现UGT84A(r = 0.85;P ≤ 2.64 × 10^-24)、F3'5'H(r = 0.55;P ≤ 4.62 × 10^-8)、DFR(r = 0.56;P ≤ 2.73 × 10^-8)和SCPL1A(r = 0.40;P ≤ 1.80 × 10^-4)基因的表达模式与儿茶素如EGCG和EGC的积累模式显著正相关,表明它们在山茶科植物中没食子酸儿茶素生物合成中的重要作用(表S13)。我们还计算了与咖啡因在山茶科植物中的积累模式相关的咖啡因相关基因的表达模式之间的相关性。结果显示,在所有研究的咖啡因合成酶基因中,IMPDA基因的表达模式与山茶科植物中咖啡因的积累模式显著相关,相关系数r = 0.77,P ≤ 6.85 × 10^-18(表S14)。类似地,观察到了GDH基因的表达模式与山茶科植物中茶氨酸积累之间的显著相关性(r = 0.56;P ≤ 2.08E-08),这可能有助于解释山茶科植物中茶氨酸的多样分布,尽管其功能还需要进一步的实验验证。我们还在山茶属物种中鉴定了SNP并进行了这些关键基因的单倍型分析,发现几个可能的单倍型与茶叶品质所需的相应代谢产物含量相关(图S3a-g)。有趣的是,我们发现F3'5'H基因在不同儿茶素含量的山茶科植物中具有高度保守性(编码序列区域的对齐一致性为98.33%),但表达水平不同。与非茶亚属植物相比,茶亚属植物的F3'5'H基因启动子克隆揭示了一个180-bp的插入,其中包含两个MYB结合位点,这可能有助于解释茶亚属植物中F3'5'H基因的高表达(图S3h-k)。此外,我们还调查了与油脂生物合成、花发育和花色素相关的关键基因在山茶科植物中的表达模式,发现只有少量与油脂代谢(28.5%)和花色素(40%)相关的基因在山茶叶片中表达较高,而大多数与花发育相关的基因在山茶叶片中表达较低(图S4)。结果还显示,在山茶叶片中,各个山茶组或物种之间没有检测到显著的差异表达模式,特别是在油茶组(富含油类代谢产物)、山茶/金花茶组(著名的观赏植物)和其他组之间(图S4)。
广泛认为,基因家族的扩张和收缩有助于代谢、调控和信号网络的动态演化(James等,2019年)。我们进一步研究了山茶科植物的基因家族扩张和收缩(表S15)。特别地,我们发现MYB、bHLH、WRKY、bZIP、MADS、AP2/ERF、GARP、NB-ARC和RLK-LRR等基因家族在茶亚属植物中过度表示,这些基因家族已被先前描述为与抗病性和植物次生代谢产物生物合成相关(夏等,2020年;杨等,2012年;赵等,2020年),与非茶亚属植物相比(图5;图S5)。我们还表明,茶亚属植物拥有更多与儿茶素、茶氨酸和咖啡因生物合成有关的结构基因的副本,包括F3'5'H、LAR、SCPL1A、GDH、ADC等(表S16)。我们还进一步检查了这些关键基因在栽培(C. sinensis)和野生(DASZ)茶树的染色体上的基因组位置模式,以确定它们是否聚集在一起形成基因簇。结果显示,与栽培和野生茶树基因组的15个假染色体上核心结构基因相关的核心基因是随机分布的。一些基因,如NMT和SCPL1A,由于串联重复事件而被扩增,没有明显的基因簇观察到(图S6和S7)。
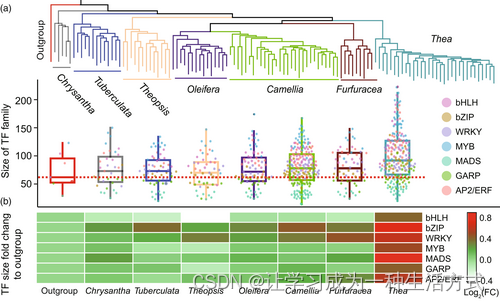
山茶科植物转录因子的扩张和收缩。(a)顶部面板显示了山茶科的系统发育,而底部面板表示了每个山茶属组的转录因子平均家族大小。(b)山茶科植物中转录因子家族的拷贝数变异。变异程度用每个组的家族大小与外类群之间的倍数变化来评估。
CTD: an updated web-based transcriptome database for genus Camellia
为了帮助研究人员更好地检索和分析转录组数据,我们发布了一个更新的基于网络的山茶科植物转录组数据库门户网站(CTD:山茶科植物转录组数据库;http://tpia.teaplant.org/transcriptome.html)。通过这个平台,用户可以广泛浏览116个山茶科植物和31个栽培茶树品种的已发布转录组信息,包括品种/物种名称、测序组织、PubMed接入号以及详细文献(图S8a)。它还提供了不同山茶科植物的busco组装完整性以及七个知名蛋白质数据库(包括Swiss-Prot、PFAM、GO、KEGG、InterPro、KOG和COG)的功能注释(图S8b、c)。此外,用户可以轻松检索和下载特定类型山茶科植物多态SSR的100-bp相邻序列和三对引物序列,这些序列可以根据缺失率、标准差和引物可转移性三种方式进行独立过滤(图S8d)。我们还提供了一个门户网站,供用户根据物种名称和TF类型轻松搜索和下载山茶科植物的70多种转录因子(图S8e)。本研究产生的所有数据,包括原始测序数据、组装转录本、功能注释、SSR和TF,都可以自由访问和下载以进行进一步分析,查询结果以用户友好的方式显示。山茶科植物和不同茶树品种的转录组数据集将持续更新,这将成为山茶科植物功能基因组学和进化生物学研究的中心入口。
DISCUSSION
High quality pan-transcriptome of 116 Camellia plants from almost all sections of genus Camellia
从转录角度来看,对山茶科植物动态系统发育、基因组演化和次生代谢产物多样化的新发现,潜在地有助于高效保护和利用山茶属种质资源,以培育栽培茶、山茶和油茶植物。尽管先前已报告了一些山茶科植物的转录组(Huang等,2017年;Shi等,2011年;Wang等,2014b年;张等,2015年),但它们在比较转录组和进化基因组研究中的质量和代表性有限。在本研究中,我们对116个山茶科植物的高质量转录组进行了测序和组装,这些植物代表了几乎所有山茶属组的物种。目前,这116个山茶植物的转录组组装总计包含了15,947,702个转录本,具有1086 bp的contig N50长度,远远长于先前报道的山茶科植物,如长叶槭茶(506 bp)(Wang等,2014b年)、油茶(771 bp)(夏等,2014年)、滇山茶(995 bp)(张等,2015年)和山茶花(996 bp)(Huang等,2017年)。整体而言,核心植物基因的广泛覆盖,加上组装转录本的高序列连续性和基因水平准确性,进一步显示了本研究中转录组组装的高完整性和准确性。基于这些山茶植物转录本,我们鉴定了大量转录因子和多态表达序列标记简单序列重复,为未来茶、山茶和油茶植物的功能基因组学和育种研究提供了宝贵的遗传资源。高质量的转录组组装还使我们初步了解了山茶植物的全转录组,进一步强化了一个单一参考转录组和/或基因组不能代表植物整体基因库的观点。总的来说,山茶科植物的转录组序列不仅显著扩展了茶植物的基因库,有利于其功能基因组学研究和遗传改良,而且为阐明山茶科植物复杂的系统发育、基因组演化和次生代谢产物的演化轨迹提供了坚实的基础数据框架。
Resolution of the phylogeny of genus Camellia
山茶属包括许多重要的植物物种(例如,茶树、油茶和山茶),在茶叶和油的生产以及观赏花卉方面起着重要作用(Chang,1981年)。然而,由于山茶属物种间频繁的种间杂交和多倍化,以及缺乏适用于进化分析的核基因,这个属的系统发育关系仍然知之甚少,这阻碍了从这个属的物种向现代育种的高效发展和利用(Xia等,2020c年)。在本研究中,我们基于405个核心低拷贝核基因解决了山茶属的系统发育问题,这是茶叶界多年来的困惑之一(Xia等,2020c年)。与先前大多数使用叶绿体(如matK、rbcL、trnL-F、rpL16和psbA-trnH)和线粒体(如matR)DNA片段构建的系统发育相比(Chen等,2014年;Yang等,2005年;Yang等,2006年;Yang等,2013年),本研究报道的山茶属系统发育受到了高度支持,大多数支系的支持率达到了100%,表明通过转录组测序获得的足够准确的低拷贝核基因对解决复杂植物群的系统发育关系具有巨大潜力。
良好解决的山茶属系统发育包括七个群,并支持了明氏分类系统(Ming&Bartholomew,2007年)中的组合,以及基于ITS和最近的核基因序列的先前研究发现(Vijayan等,2009年;Zhang等,2022年)。通过Camellia植物的表型数据(Hong,2011年;Lu等,2012年),特别是在一些重要和复杂的支系(表S17)中,也支持了构建的山茶属系统发育的准确性。例如,山茶属植物中的茶和无毛亚组在整个叶片形状、网状叶脉和叶缘形状及锯齿方面具有相似的表型特征,这表明了它们在构建的系统发育树和次生代谢物分布方面的分类组合。类似的叶片构造特征也观察到了油茶和山茶亚组植物之间,包括类似的叶缘形状和锯齿,以及次级叶脉和主脉之间的夹角,支持了它们在系统发育树中的密切关系。同样,疏花亚组和伪山茶亚组(例如顶部、无毛的背面、次级叶脉形状和小叶组成)以及铁线莲亚组和山茶花亚组(例如整个叶片形状、顶部、叶缘形状和锯齿,以及小叶组成)之间也呈现出类似的形态特征,这与它们在系统发育树中的密切关系一致。本研究构建的系统发育树不支持以前报道的基于ITS和核基因的树中提出的山茶亚属的多源性特性(Vijayan等,2009年;Zhang等,2022年)。我们的转录组基础树显示,山茶亚属在我们的系统发育树中是单源性的,这与形态数据的发现一致(Lu等,2012年;明,1998年)。此外,与先前基于ITS的树相比,本研究构建的基于转录组的树显著提高了山茶属系统发育树的分辨率和可靠性。例如,Thea、Eriandra、Theopsis和Camellia亚属显示出梳状的拓扑结构,在ITS基础树中无法很好地区分,而在基于转录组的树中,所有这些亚属的物种都被明确分类并得到了高支持值。用于构建系统发育树的有限信息位点可能是导致ITS基础树分辨率和可靠性低的主要原因。此外,结果还显示了山茶属植物核基因组和叶绿体基因组的系统发育树之间的不一致性(图S10和S11)。此外,我们估计山茶植物起源于约14.30百万年前,Thea亚属植物的分化发生在约6.67百万年前。特别是,栽培茶植物的两个变种的分化时间可能起源于0.82-2.16百万年前,这与第四纪更新世的更新世时期一致,与使用小部分共线基因估计的0.38-1.54百万年前(Wei等,2018年)一致,但晚于使用全基因组核基因数据集估计的时间(Xia等,2020a年)。
Genome evolution of Camellia plant associated with its phenotypic diversification
先前的研究已经证实,栽培茶树(C. sinensis)和许多其他开花植物一样,经历了一个约3,000-4,000万年前的近期全基因组重复事件(WGD)(Wang等,2021年;Xia等,2017b年);然而,目前尚不清楚这个WGD事件是否发生在其他山茶属植物身上,以及它如何影响山茶植物的多样化和品质特征。我们的研究结果提供了补充证据,表明栽培茶树、野生茶树和油茶植物等山茶属植物的最近共同祖先与猕猴桃等番木瓜目植物共享了一个近期的WGD事件,通过基因组共线性分析得到了证实(Gong等,2022年;Xia等,2020b年;Zhang等,2020b年)(图S1d)。对代表性山茶植物,包括栽培茶树(C. sinensis)、油茶树(C. oleifera)和常见山茶(C. reticulata)的WGD衍生复制基因进行功能注释后发现,这些WGD事件衍生的重复基因中的一些与UDP-糖基转移酶活性、脂质生物合成过程、芳香化合物生物合成过程和色素代谢过程相关。这使我们得出结论,最近的WGD事件很可能在建立山茶植物的农艺性状和多样化表型特征方面发挥了关键作用。
Transcriptomic mechanism underlying diverse distribution of tea-quality related characteristic secondary metabolites in Camellia plants
我们在一个良好解决的系统发育框架下提供了山茶植物次生代谢产物的进化景观。我们观察到在山茶属植物的Thea组中,特别是栽培茶树中,茶叶品质相关的次生代谢产物,包括儿茶素、咖啡因和茶氨酸的显著积累,这很可能是人工和自然选择的结果,并且与先前报道的Thea组物种相似(Xia等,2017a年;Xia等,2019年)。值得注意的是,某些复杂的山茶属植物亚科,如Glaberrima、Thea、Tuberculata和Pseudocamellia组的系统发育关系也得到了次生代谢产物分布模式的支持。这不仅表明了使用核基因构建的系统发育树的可靠性和稳健性,还暗示次生代谢可以作为未来山茶植物分类的一个参数。我们的结果还显示,在山茶植物中,儿茶素、咖啡因和茶氨酸的积累之间呈正相关,这与先前的研究结果一致,显示出这三种代谢物的代谢可能在茶树中共同调节(Tai等,2018年)。我们发现与抗病性相关的基因家族(如NBS-LRR和RLK-LRR)在Thea组物种中扩展,而几个与茶叶品质相关的基因(如F3’5’H、DFR、SCPL1A和UGT84A)在山茶植物中的积累模式中表现出很高的相关性,可能有助于茶叶品质的形成和山茶植物的适应性(Xia等,2017a年;Xia等,2020a年)。有趣的是,我们发现,尽管CsTSI是栽培茶树中茶氨酸生物合成的关键基因(Wei等,2018年),但山茶植物中茶氨酸的积累与CsTSI基因的表达并没有显著相关,而与AlaDC和GDH基因的表达呈显著正相关。这表明在TSI的存在下,AlaDC和GDH可能是茶氨酸生物合成的两个枢纽基因,提供了在许多先前研究中揭示的关键底物(乙胺和谷氨酸)(Cheng等,2017年;Yang等,2020年;Zhu等,2021年)。未来还需要进行功能实验,进一步确定这些与品质相关的候选位点和基因组调控元素如何参与山茶植物的表型特征建立和生态适应。此外,我们建立了一个全面的网络可访问数据库,帮助研究人员更有效地利用山茶植物的转录组进行茶叶、山茶和油茶植物育种,这也将成为未来山茶植物功能基因组学和进化生物学研究的中心门户。
总之,我们提供了116个山茶植物的高质量转录组,并对它们的深层系统发育和与环境适应性和品质特征相关的基因和基因组进化提供了新的见解。我们还确定了与茶叶品质相关的三种主要次生代谢产物的含量,并在一个良好解决的系统发育框架下调查了它们的进化轨迹和基因表达模式。本研究的转录组序列和总体发现不仅有助于山茶属植物的比较转录组学和功能基因组学研究,还为未来改良饮用茶、油茶和常见山茶植物提供了宝贵的资源,以满足全球消费者日益多样化的需求。
EXPERIMENTAL PROCEDURES
样品收集、文库构建和RNA测序
2018年春末,从浙江省金华国际山茶物种园收集了山茶植物和Polyspora speciosa的新鲜健康幼嫩叶片。收集后,所有样品立即冷冻在液氮中,并在RNA提取和次生代谢物评估之前在-80°C保存。使用改良的溴化十六烷基三甲基铵法从每个山茶植物和P. speciosa的幼嫩叶片中提取总RNA,并使用DNase I进行处理(Xia等,2017a)。在通过Agilent 2100 Bioanalyzer(Agilent Technologies, Santa Clara, CA, USA)进行完整性检查后,得到的高质量RNA被用于按照Illumina的标准方案构建测序文库,并使用Illumina Novaseq平台进行配对末端测序策略进行进一步测序。对RNA测序进行了三次生物复制。我们还从NCBI数据库收集了9个栽培茶树品种的原始RNA测序数据(表S1)。使用Trimmomatic对生成的原始测序reads进行预筛除低质量碱基(phred得分<20)、连接器、重复和潜在污染(Bolger等,2014年)。
新组装和完整性评估 使用默认参数将干净的测序reads进行新组装成转录本(Haas等,2013年)。通过cd-hit处理产生的转录本以减少组装过程中引入的不可避免的冗余(Fu等,2012年)。还删除FPKM值低于1的转录本,以避免潜在的组装错误。使用busco评估转录组组装的完整性,使用植物数据集(Simão等,2015年)。
这个删除过滤有意思了,工作量也有,又表明了真实可靠。
功能注释、SSR和TF鉴定
通过将山茶转录本与七个公共知名蛋白数据库(包括Swiss-Prot、PFAM、GO、COG、KEGG和拟南芥(TAIR10)数据库)进行比对,使用blastall进行功能注释,E≤1×10^–5(Altschul等,1997年)。使用misa工具识别山茶转录本中的SSR,使用默认设置(Beier等,2017年)。进一步使用CandiSSR管道识别不同山茶植物中的多态性SSR(Xia等,2015年)。使用itak工具进行山茶TF的预测和分类,使用默认参数(Zheng等,2016年)。
同源基因鉴定和低拷贝基因选择
采用类似于之前描述的方法(Zeng等,2014年)进行了山茶植物中低拷贝同源基因的鉴定。简而言之,首先使用OrthoFinder管道构建了山茶植物内/间的推测同源和同源基因组,采用默认参数(Emms&Kelly,2019年)。然后使用内部perl脚本提取了低拷贝同源基因对。为了进一步获取用于构建系统发育树的高质量单拷贝基因,我们构建了每个低拷贝同源基因组的单基因树,并手动检查了单基因树以删除产生“长枝吸引”现象的分类单元。此外,如果在同一分类单元中存在两个以上的同源基因副本,则删除长度较短的基因,以获取最终用于构建系统发育树的单拷贝同源基因组。
系统发育树构建和分化时间估计
为了构建山茶植物的系统发育树,首先使用默认参数将每个单拷贝基因同源的氨基酸序列进行了比对(Edgar,2004年)。然后,使用trimal对每个同源基因的比对文件进行修剪,以去除不良比对区域(Capella-Gutierrez等,2009年)。基于修剪和串联的单拷贝基因比对,使用P. speciosa作为外类群,使用raxml构建了山茶植物的系统发育树(Stamatakis,2014年)。使用prottest估计了用于构建系统发育树的最佳适配模型Jones–Taylor–Thornton模型(即JTT模型)(Darriba等,2011年)。引导重复设置为1000。使用astral构建了属山茶植物的共同祖先系统发育树(Zhang等,2018年)。使用r8s程序估计了不同山茶植物系之间的分化时间(Sanderson,2003年)。山茶属与多肉植物的分化时间(14.50百万年前)被用作校准点(Li等,2013年)。
山茶植物中WGD事件的鉴定
使用wgd管道鉴定了代表性山茶植物中发生的WGD事件,该管道实施了用于构建Ks分布的方法、用于基因组内句法分析的工具以及用于建模和可视化Ks分布的方法(Zwaenepoel&Van de Peer,2019年)。简而言之,首先使用blastall对每个代表性山茶植物预测的编码序列进行全对比(Altschul等,1997年)。然后使用马尔可夫聚类算法对比对结果进行聚类以产生全基因组句法(Van Dongen,2000年)。从每个山茶植物的句法中提取编码序列,并使用fasttree推断了系统发育树(Price等,2010年)。然后,为了推断WGD事件,从系统发育树识别出的同源家族中生成Ks分布,使用wgd软件包中实现的ksd命令(Zwaenepoel&Van de Peer,2019年)。
儿茶素、茶氨酸和咖啡因含量的确定
我们使用HPLC确定了与茶叶品质相关的三种次生代谢产物——儿茶素、茶氨酸和咖啡因的含量。简而言之,将粉碎的真空冷冻干燥叶片(0.5g)用含有80%甲醇、1%醋酸和19%纯水的提取溶液(在15°C超声波处理10分钟并搅拌)进行提取,然后在室温下以3420g离心10分钟。将上清液取出至10mL容量瓶中,再用提取溶液稀释至10mL。将提取液通过0.22μm有机相Millipore滤器(MilliporeSigma,Burlington,MA,USA)过滤,用Agilent 1260系列HPLC系统进行儿茶素、没食子酸(GA)和咖啡因的分析,包括样品管理器、四元溶剂管理器、紫外/可见检测器和NX-C18柱(粒径5μm,柱尺寸250mm×4.6mm;Gemini;Phenomenex,Torrance,CA,USA)。色谱测定方法按照先前的描述进行(Mao等,2018年)。对于茶氨酸的测定,将粉碎样品(0.05g)用热水(100°C)浸泡20分钟,然后通过0.22μm水相Millipore滤器进行过滤,用Agilent 1260系列HPLC系统和HSS-T3柱(粒径5μm,柱尺寸250mm×4.6mm)进行分析。色谱测定方法按照中国国家标准GB/T 23193–2017(茶叶中茶氨酸的测定—使用高效液相色谱法)进行。
参考文献
Comparative transcriptomic analysis unveils the deep phylogeny and secondary metabolite evolution of 116 Camellia plants
Qiong Wu, Wei Tong, Huijuan Zhao, Ruoheng Ge, Ruopei Li, Jin Huang, Fangdong Li, Yanli Wang, Ali Inayat Mallano, Weiwei Deng, Wenjie Wang, Xiaochun Wan, Zhengzhu Zhang, Enhua Xia
First published: 05 May 2022

















 1751
1751

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









