










Integrated multi-omic data and analyses reveal the pathways underlying key ornamental traits in carnation flowers
综合多组学数据和分析揭示了康乃馨花卉关键观赏性状的底层途径,康乃馨基因组,致敬520~

摘要
康乃馨(Dianthus caryophyllus)是世界上最受欢迎的观赏花卉之一。尽管已有众多关于康乃馨的研究,但花的颜色、香气以及复瓣花的形成机制尚不明确。在这里,我们采用了一种综合多组学方法来阐明康乃馨花卉最重要的观赏性状的遗传和生化途径。首先,我们组装了一个高质量的染色体级康乃馨基因组('Scarlet Queen',636Mb,contig N50为14.67Mb)。接下来,通过多种仪器类型从花的不同部位在多个发育阶段生成了一系列代谢组数据集,以评估色素和挥发性化合物积累的空间和时间差异。最后,生成了转录组数据,将基因组、生化和形态学模式联系起来,提出了一套形成花瓣着色、复瓣花和香气产生等观赏性状的途径。其中,转录因子bHLHs、MYBs和WRKY44同源体被认为在控制花瓣颜色图案形成中很重要,而像松香醇乙酰转移酶和丁香酚合成酶这样的基因则参与了丁香酚的合成。这里呈现的基因组学、转录组学和代谢组学的综合数据集为理解花的发育和着色的底层途径提供了重要的基础,进而可以用于选择性育种和基因编辑,以培育新的康乃馨品种。
引言
康乃馨(Dianthus caryophyllus L.,2n=2x=30)是全球最重要的观赏花卉之一,仅次于玫瑰,非常受欢迎。康乃馨具有高经济和文化价值,被广泛用于多种观赏用途,包括鲜切花、盆栽和园林绿化。康乃馨的栽培历史超过2000年。康乃馨花常用于纪念多种仪式、事件和节日,如中国的婚礼庆典、法国的宴会和美国学生的舞会。植物育种家利用种间杂交开发了具有新颖花卉和生长特性的品种(Nimura等,2006;Sparnaaij等,1990)。
多样化的花卉形态和香气吸引了千年来的人类,这在日常生活中花卉的无数审美用途中得到了体现。颜色是人类在评价花卉美学品质时最重要的特征之一。康乃馨品种的花色非常多样,包括红色、粉色、白色、洋红色和黄色,以及同一花瓣上多种颜色的各种颜色图案。由于新颖的颜色和/或图案非常受欢迎,可以增加销售额,研究人员不断尝试开发新颖有趣的品种。以前的研究报告了纯色系列康乃馨的分子机制,包括白色(Onozaki等,1999)、粉色(Mato等,2001)和黄色(Itoh等,2002;Yoshida等,2004)。基于这些研究,使用遗传工程技术开发了一种新的蓝色康乃馨(Fukui等,2003;Matsuba等,2010)。育种新的康乃馨品种将吸引更多的关注并产生高经济价值。然而,急需了解多色花的底层分子机制。
花卉形态是消费者最喜爱的特征之一,也是康乃馨研究者关注的重要话题。在拟南芥中著名的ABC模型(Coen和Meyerowitz,1991)和“花卉四重奏”模型(Pelaz等,2000;Theißen和Saedler,2001)表明,A类、A+B类、B+C类和C类基因参与了萼片、花瓣、雄蕊和心皮的发育。由于双花表型最受欢迎,因此使用来自单花亲本和双花亲本的F2代子代构建了遗传连锁图,从而确定了关键调控因子(如APETALA2-like(AP2L)基因)(Wang等,2020a;Yagi等,2013)。康乃馨的辛辣香味也被视为一种理想特性,其中的主要成分是丁香酚(Clery等,1999;Kishimoto等,2011;Onozaki,2018)。由于其非凡的香气,康乃馨获得了中文名“香石竹”,其中“香”代表香气,“石竹”指的是属名Dianthus。之前的研究大大增进了我们对康乃馨关键观赏性状的了解(Yagi等,2013、2014、2017;Zhang等,2018),但当前可用的基因组是一个包含45,088个scaffold的草稿组装,需要改进。了解关键性状背后的分子机制并将这些知识应用于通过标记辅助育种和基因编辑开发改良品种非常重要。
在此,我们基于来自牛津纳米孔技术(ONT)的PromethION平台生成的序列数据,以及使用高通量染色体构象捕获(Hi-C)技术构建的假染色体,提供了一个组装和注释的高质量康乃馨品种‘Scarlet Queen’参考基因组。基于这个参考基因组,通过全面的同源性搜索、基因家族分析、代谢组数据的整合以及来自RNA-seq数据集的共表达分析,确定了康乃馨重要观赏性状的遗传调控因素,包括花瓣边缘和边缘(技术上是顶端的边缘;此后为简化称之为‘边缘’)的着色、丁香酚生物合成以及花瓣状雄蕊的形成。这项工作提供了开发康乃馨改良和新颖观赏性状所需的高质量基因组资源。
结果
康乃馨高质量染色体级基因组的组装与注释
为了全面促进康乃馨观赏性状分子机制的分析,我们生成了一个高质量的康乃馨基因组。2014年发布的之前的康乃馨基因组(‘Francesco’_r1.0,美国国家生物技术信息中心(NCBI)登录号GCA_000512335.1)是一个草稿组装,它仅使用Illumina和GS FLX+短读技术生成,结果产生了45,088个scaffold,覆盖568.9Mb,scaffold N50为60.73kb(Yagi等,2014)。在本研究中,我们生成了95倍覆盖度的ONT 60.65Gb pass读取,并组装了D. caryophyllus ‘Scarlet Queen’(一种以花瓣边缘着色著称的品种)基因组(图1a)。Illumina短读(45.41Gb ~62.60倍覆盖度,过滤后)被用来进一步优化ONT数据(表S1和图S1)。由于其1.2%的杂合性率(图S2和S3及表S2),从最初组装的基因组中移除了冗余序列,以获得一个去冗余的基因组。最终的636.30Mb基因组组装(D. caryophyllus v1.0)仅包含75个contig,contig N50为14.67Mb,接近通过K-mer调查和流式细胞仪获得的预计基因组大小约640Mb(表S2和图S4)。Hi-C的配对末端读取被用来聚类、排序和定向(表S3),然后使用LACHESIS包与手工调整,将580.05Mb(91.16%)锚定到15个假染色体上(2n = 30)。最终组装达到N50为38.55Mb,假染色体长度从30.95Mb到53.54Mb不等(表1,图1b和图S5)。基准普遍单拷贝直系同源基因(BUSCO)分析结果显示完整性达到了97.15%(表S4)。长末端重复(LTR)-组装指数为12.11。我们重新映射并获得了Illumina(99.77%)和ONT(99.77%)数据的高映射率(表S5-S7)。使用‘72L’(Yagi等,2017)的康乃馨遗传图来评估组装基因组的质量。结果显示‘Scarlet Queen’的组装基因组与‘72L’的遗传图之间具有更高的同线性(平均91.47%)(图S6)。以上所有提到的基因组指标表明,为康乃馨新组装的基因组质量很高。
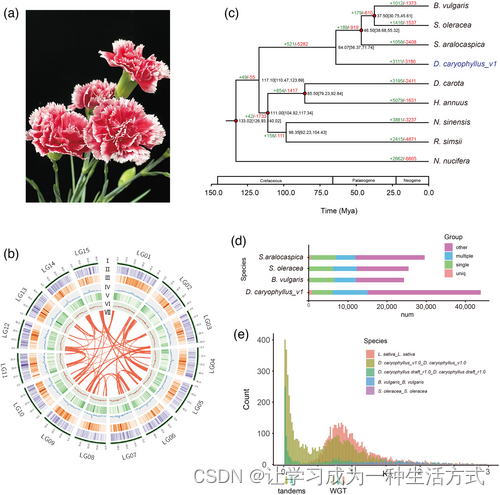
康乃馨D. caryophyllus基因组的系统发育分析。 (a) D. caryophyllus ‘Scarlet Queen’的花。 (b) 康乃馨基因组特征分布。15个假染色体的圆形表示(I)染色体长度(以Mb为单位),(II)基因密度,(III)所有LTRs的密度,(IV)所有Gypsy-LTRs的密度,(V)所有Copia-LTRs的密度,(VI)500kb窗口中的GC含量,以及(Ⅶ)圆心的每条连接线连接一对同源基因。 (c) D. caryophyllus与其他八个双子叶植物种的系统发育树。基因家族扩张用绿色表示,收缩用红色表示。每个节点处标示的估计分化时间(百万年前,MYA),括号中的数字是95%置信区间(每个中心定义为平均值)。红点表示一个校准点。 (d) D. caryophyllus_v1及其他已测序基因组的不同组中的基因显示,值用条形图显示。 (e) Ks值显示与S. oleracea和B. vulgaris相比,康乃馨发生了一次多倍体事件,这两者近期未经历多倍体事件,而L. sativa则经历了近期的三倍体事件。
| ‘Scarlet Queen’ | ‘Francesco’ | |
|---|---|---|
| Sequencing | ||
| ONT | ~60.65G | * |
| Illumina-Short | ~48.86G | * |
| Illumina-HIC | ~81.70G | * |
| Assembly | ||
| Contig N50 | 14.67 Mb | 17.55 kb |
| Contig N90 | 4.53 Mb | 2.68 kb |
| scaffolds N50 | 38.55 M | 62.62 kb |
| scaffolds N90 | 30.95 M | 8.0 kb |
| Longest contig | 34.83 Mb | 1.29 kb |
| Number of contigs | 75 | 88,654 |
| Number of scaffolds | 61 | 45088 |
| De-redundancy total | 636.30 Mb | 568.89 Mb |
| BUSCO | 97.15% | – |
| Total GC content (%) | 38% | 36% |
| TE | 70.62% | 53.96% |
| Heterozygotic | 1.2% | 0.2% |
| Annotation | ||
| Number of predicted genes | 43925 | 43266 |
| Swiss-prot | 21700 (49.4%) | * |
| Interproscan | 37656 (85.73%) | * |
| GO | * | 16423 (37.8%) |
| NR | 21133 (48.11) | * |
| KOG | 13896 (31.64%) | 19005 (43.7%) |
| Annotated | 40332 (91.82%) | * |
| Unannotated | 3593 (8.18%) | * |
| BUSCO | 95% | 82.1% |
共预测出43,925个编码蛋白的基因(表S8和S9以及图S7)。最终,91.82%的预测基因使用不同数据库进行了注释(表S10)。从康乃馨基因组注释的基因数量显著多于菠菜(Spinacia oleracea)、碱蓬(Suaeda aralocaspica)和甜菜(Beta vulgaris)(图1d)。此外,使用Infernal预测出1,453个转移RNA、594个核糖体RNA(rRNA)、2,433个小核RNA和95个微小RNA(mRNA)基因(表S11)。对新组装基因组的BUSCO分析显示,完整性为95%,而之前的D. caryophyllus_r1草稿基因组(Yagi等,2014)使用相同流程(Embryophyta_odb10数据库)的完整性仅为82.1%(表1),这表明我们的基因组具有更高的完整性。在我们的康乃馨基因组中,重复序列占70.62%,其中LTR逆转录转座元素占基因组的36.14%,包括9.03%的copia型和剩余的27.11%的gypsy型(图1b、表S12和S13以及图S8)。被称为‘Scarlet Queen’的康乃馨基因组高度杂合。因此,我们使用之前发表的方法(Zhong等,2021)检查了我们新组装基因组内的序列多态性。我们发现1.79百万单核苷酸多态性(SNP),其中1.15百万是转换,0.74百万是颠换(表S14和S15,图S9-S11)。36.96%的SNP位于基因间区域。SNP导致2044个基因的起始或终止密码子的丢失/获得。我们进一步识别出0.53百万个插入/删除(indel),其中598个导致起始或终止密码子的丢失或获得(表S14和S15)。
进化和全基因组复制
我们构建了一个系统发育树,并使用OrthoFinder识别的1433个共享单拷贝基因估计了九种植物的分化时间(图1c)。结果显示D. caryophyllus与藜科的物种(例如菠菜、甜菜和碱蓬)关系最为密切。使用不同的校准点估计康乃馨和藜科的分化时间约为6407万年前(MYA)(图1c和S12)。在D. caryophyllus基因组中,3111个基因家族发生了扩张,3180个基因家族发生了收缩(图1c和S13)。通过基因本体(GO)富集分析,扩张的基因家族在RNA糖苷酶活性、rRNA N-糖苷酶活性、ADP结合、水解酶活性、水解N-糖苷化合物、过渡金属离子结合、催化活性作用于rRNA、锌离子结合和核酸结合等方面富集(表S16)。此外,与未经历近期多倍体事件的菠菜和甜菜的同义替换率(Ks)以及经历了近期全基因组三倍体事件(WGT)的莴苣(Lactuca sativa)的Ks和4dTv值表明,康乃馨可能经历了一次全基因组多倍体事件(图1e和S14)。D. caryophyllus_v1与葡萄(Vitis vinifera)、甜菜和菠菜之间的同线性模式为3:1(图S15-S21)。Ks值、4dTv值和同线性模式的结果表明,康乃馨在石竹科与藜科分化后可能经历了一次全基因组三倍体事件。
花瓣主要色素的鉴定和定量
在这项工作中,我们采集了‘Lorca’的组织样本,该品种具有红色边缘和黄色花瓣花瓣,以阐明花瓣色素在空间和时间上的差异(图2a)。选择了三个不同阶段的花瓣进行代谢物检测:第3阶段(完全闭合的花,花瓣无红色),第4阶段(花瓣开始变红),第6阶段(开放的花,花瓣呈红色)。
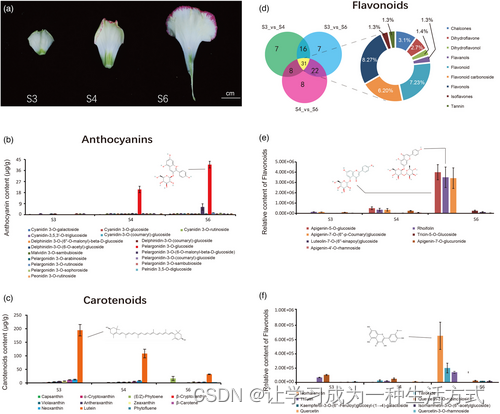
康乃馨花瓣色素的鉴定和定量。 (a) 三个阶段,S3:花瓣无颜色,S4和S6:花瓣着色。这些花瓣被用于代谢物检测。 (b) 从S3、S4和S6阶段的花瓣中鉴定并定量了18种花青素。 (c) 从S3、S4和S6阶段的花瓣中鉴定并定量了11种类胡萝卜素。 (d–f) 从S3、S4和S6阶段的花瓣中鉴定并定量了黄酮类化合物。 (d) S3对S4、S3对S6和S4对S6之间差异积累代谢物的文氏图。 (e) 不同阶段七种高丰度黄酮类化合物的数量。 (f) 不同阶段八种高丰度黄酮醇的数量。 通过液相色谱-电喷雾电离串联质谱(LC-ESI-MS/MS),在花瓣中鉴定并定量了30种花青素化合物(针对99种花青素标准)(图S22和表S17)。通过比较不同发育阶段的这些花青素(图2b显示前18种),我们发现S6阶段的花瓣中洋牡丹素3-O-葡萄糖苷含量最高,其次是洋牡丹素3-O-(6-O-马洛尼基-beta-D-葡萄糖苷)(图2b)。鉴于这两种化合物的增加与花瓣发育和边缘更深的红色相关,我们认为这些花青素是花瓣边缘红色着色的主要化合物。通过超高效液相色谱(UPLC)分离,定量了不同阶段花朵中的类胡萝卜素含量(补充说明和图S23)。在检测到的类胡萝卜素中,叶黄素含量在三个阶段中均为最高(图2c和表S18)。尽管叶黄素是最丰富的类胡萝卜素,但随着花的发育其含量减少。植物烯在S6阶段增加,但其浓度仍低于叶黄素。此外,我们还使用UPLC在三个花瓣阶段检测并定量了184种黄酮类化合物(表S19和图S24)(补充方法说明)。通过我们的比较分析(S3对S4、S4对S6和S3对S6),黄酮类化合物含量的差异(倍数变化≥2,倍数变化≤0.5,以及投影得分变量重要性≥1)在每个花发展阶段都有所不同;然而,31种黄酮类化合物在所有比较组中共有,主要共有化合物是二氢黄酮(数量=8,占27%)、黄酮类(7,23%)和二氢黄烷酮(6,20%)(图2d和表S20)。在黄酮类化合物中,S6阶段的黄芩素-5-O-葡萄糖苷、染料木黄酮和黄芩素-7-O-(6″-p-香豆素)-葡萄糖苷含量显著高于S3和S4阶段(图2e)。在黄酮醇中,异鼠李素在S6阶段最为丰富。这些含量可能有助于‘Lorca’花瓣从白绿色到黄色的着色过程(图2f)。
花瓣颜色中涉及的色素合成途径基因的表征
为了进一步了解色素生物合成,特别是花青素生物合成途径,我们采集了七个不同阶段(S1-S3样本显示无色花瓣,S4-S7阶段花瓣显示红色边缘,其中‘Lorca’花瓣在S4阶段逐渐形成红色边缘)的样本以提取和测序基因转录物。
基于同源性搜索和基因组功能注释,我们识别了多个涉及色素生物合成的基因,包括PAL、C4H、4CL、CHS、CHI、F3H、FLS、DFR、ANS和AT/GT。我们根据京都基因与基因组百科全书数据库以及之前为其他高等植物发布的数据(Belwal等,2020;Falcone Ferreyra等,2012),勾画出了假设的色素生物合成途径(图3)。我们发现,在S4至S7阶段,DFR和ANS的mRNA水平高于前三个花发展阶段(图3a)。Dca_v1_10 g.0063700_FLS也观察到类似的表达模式。在众多植物物种中已描述了类胡萝卜素生物合成的发生(Sun等,2018)。根据功能注释,我们在康乃馨中识别了两个PSY、三个PDS、五个ZDS/CRTISO、两个LCYB/LCYE、一个CHYB、八个ZEP和两个VDE类胡萝卜素生物合成基因(图3b)。随着花瓣的发展,被认为是类胡萝卜素骨架产生的限速酶的Dca_v1_ 13g.0238600_PSY的表达减少。其他下游基因,如Dca_v1_07g.0027500_PDS、Dca_v1_08g.0280000_LCYE、Dca_v1_01g.0015600_ZEP、Dca_v1_10 g.0104200_ZEP和Dca_v1_15g.0018200_ZEP的同源基因也显示了相同的表达模式,导致类胡萝卜素积累减少。这些结果对理解色素积累和着色特别感兴趣。
色素生物合成途径。 (a) 提议的花青素和黄酮醇的合成途径。 (b) 提议的叶黄素和类胡萝卜素的合成途径。苯丙氨酸解氨酶(PAL)、肉桂酸-4-羟化酶(C4H)、4-香豆酸辅酶A连接酶(4CL)、查尔酮合成酶(CHS)、查尔酮异构酶(CHI)、黄烷酮3-羟化酶(F3H)、二氢黄烷酮4-还原酶(DFR)、花色苷合成酶(ANS)、酰基转移酶(AT)、葡萄糖转移酶(GT)、黄酮醇合成酶(FLS)、植物烯合成酶(PSY)、植物烯去饱和酶(PDS)、类胡萝卜素异构酶(CRTISO)、番茄红素β-环化酶(LCYB)、番茄红素ε-环化酶(LCYE)、细胞色素P450类胡萝卜素β-羟化酶(CYP97A)、细胞色素P450类胡萝卜素ε-羟化酶(CYP97C)、玉米黄质环氧化酶(ZEP)、紫黄质脱环氧化酶(VDE)和新黄质合成酶(NXS)。不同阶段基因表达谱(FPKM),表达量由低到高用颜色变化从蓝色(低积累)到红色(高积累)表示。
MYBs、bHLHs和WRKYs参与花瓣边缘的着色
为进一步澄清参与花瓣边缘红色着色的基因,我们进行了差异表达基因(DEGs)的比较分析,分析了代表花瓣颜色变化主要阶段的两组合并样本(边缘对比花瓣和S3对比S4)(图4a)。在这两个比较中,分别有4140(S3对S4)和626(边缘对花瓣)的特异DEGs(图4b和表S21、22)。在康乃馨花发展期间,我们鉴定了两组之间重叠的275个DEGs。DEGs的GO富集分析表明,这些基因富集于氧化还原酶、水解酶(作用于糖基键,水解O-糖基化合物)和转录调控活性以及其他代谢和生物过程(图4c)。以前的研究表明,MYBs和bHLHs参与花青素生物合成(Xu等,2015)。我们通过分析它们的系统发育和基因结构(图S25)以及位置(图S26),鉴定了MYB家族,并分析了来自275个DEGs的MYBs和bHLHs的表达(图S27和S28)。结合特征性状,鉴定了九个与红色边缘着色相关的候选基因(图4d和S29)。其中,我们发现FLS、DFR、ANS、两个MYBs和WRKY44的表达模式与花的发展相似。这些基因的表达随着红色加深而增加,然后减少;这些基因在花瓣边缘的表达量高于花瓣中部。通过检查基于WGCNA(一个R包)推断的共表达网络,我们构建了加权基因共表达网络并获得了30个簇(图S30和S31)。此外,基于与花青素合成相关的基因,构建了一个调控网络(图S32)。上述九个候选基因的调控网络也被提出(图4e),这表明MYBs、bHLHs和WRKY44 TFs参与了花青素合成。为全面检查参与花青素/黄酮醇生物合成途径的基因调控,提出了一个关于康乃馨花瓣红色边缘形成的调控网络(图4f);当Dca_v1_10g0111900_MYB基因的表达在花瓣边缘与其他MYBs、bHLHs和WRKY44一起特异性上调时,它促进了下游DFR和ANS表达的增加,从而有助于红色边缘的形成。
花瓣边缘着色的形成。 (a) 不同发育阶段的花瓣(花冠裂片)形态和着色。 (b) 文氏图显示两次比较中差异表达基因(DEGs)的数量。 (c) 两次比较中DEGs的基因本体(GO)富集分析。 (d) 7个阶段和两种组织中花青素合成相关基因的表达。 (e) Dca_v1_10g0111900_MYB的子网络,数值低则圆圈小。从低到高的表达通过颜色从蓝色变为红色来表示。 (f) 提出的花瓣着色的调控途径。
康乃馨中丁香酚合成的提议生物合成途径
我们发现三种主要挥发性物质,即顺式-3-己烯基苯甲酸酯、β-香叶烯和丁香酚,是S6阶段康乃馨花香的主要成分,通过气相色谱-质谱(GC-MS)分析得出(图5a)。丁香酚通常被认为是康乃馨的特征香气。为了进一步评估丁香酚的生物合成,测定了各个花发育阶段(S2–S6)花中的丁香酚稳态水平。结果显示丁香酚含量随花的发育而变化。具体来说,从S4阶段的0.00708毫摩尔/克增加到S5阶段的0.02596毫摩尔/克,以及S6阶段的0.04247毫摩尔/克(图5b)。特别值得注意的是,丁香酚和花青素的积累都从S4阶段开始,并在之后呈现类似的积累模式。
康乃馨中丁香酚合成的提议生物合成途径。 (a) 通过固相微萃取气相色谱-质谱(SPME-GC_MS)鉴定了三种挥发性物质。 (b) 不同花发育阶段的丁香酚含量。 (c) 康乃馨中丁香酚合成的提议生物合成途径。苯丙氨酸解氨酶(PAL)、肉桂酸-4-羟化酶(C4H)、查尔酮合成酶(CHS)、查尔酮异构酶(CHI)、黄烷酮3-羟化酶(F3H)、二氢黄烷酮4-还原酶(DFR)、花色苷合成酶(ANS)、UDP-黄酮苷基转移酶(UFGT)、咖啡酰辅酶A-O-甲基转移酶(CCOMT)、肉桂酰辅酶A还原酶(CCR)、肉桂醇脱氢酶(CAD)、松香醇乙酰基转移酶(CFAT)、丁香酚合成酶(EGS)。
为了解析丁香酚的合成途径,我们结合康乃馨基因组和转录组数据,鉴定了与丁香酚合成相关的34个基因,包括三个CCOMT基因、十一个CCR、九个CAD、六个松香醇乙酰基转移酶(CFAT)和五个丁香酚合成酶(EGS)基因。其中,上述提到的扩展基因包含了属于锌离子结合GO项,参与丁香酚合成途径的CFAT基因。我们还发现,CFAT基因Dca_v1_12g.0191300和EGS同源基因Dca_v1_10g.0152400从S4阶段开始增加,这与丁香酚开始积累的时间相对应(图5c)。在‘Lorca’(丁香酚含量丰富)和‘Francesco’(丁香酚含量不丰富)品种的EGS同源基因的系统发育分析、序列比对和启动子元素比较中观察到几个重要差异(Kishimoto,2020)(图S33)。在‘Francesco’的Dca27403中发现了一个在最后一个外显子中的提前终止密码子,使得翻译的蛋白质缩短了15个氨基酸。在‘Lorca’中Dca_v1_10g.0152400的启动子与‘Francesco’中的Dca36983和Dca27403基因相比,含有较少的调节元素。
复瓣康乃馨ABC基因的表达模式
为了表征花的启动和发育的遗传模式,选择了七个阶段进行分析,包括S1:活跃的腋芽,茎顶端分生组织转化为花原基,S2:花分生组织和早期花器官发育,S3:完全闭合的花,S4:开始着色的花,S5:开花前的花苞和着色,S6:开放的花,S7:衰老的花(图6a)。
花的发育基因基础。 (a) 花的发育阶段(S1-S7)。 (b) 类型Ⅱ MADS-box家族的系统发育树,康乃馨基因以蓝点表示。 (c) 不同花器官中A-/C-功能基因的表达模式。Pe-out (A):外层花瓣;Pe-mid (B):内层花瓣;Pel-st (C):花瓣状雄蕊;St (D):雄蕊。 (d) 在拟南芥中35S::DcaPI2.2的表型。35S::DcaPI2.2转基因花展开(ⅰ)并包含花瓣状萼片(ⅱ),Col表示哥伦比亚拟南芥。 (e) 康乃馨复花表型中的ABC模型,其中A类基因表达在各花器官中保持不变,导致内轮变为花瓣化。S:萼片;P:花瓣;PI:花瓣状雄蕊;St:雄蕊;C:心皮。
通过检查上述共表达网络并结合基因组的注释信息,我们发现深灰和花白模块与开花的启动有关,古白、淡绿松石和棕色模块与花器官的发育有关,而深橄榄绿模块与衰老有关(图S34)。因此,这些模块的识别有助于分析这些生物过程。基于在拟南芥中报道的花发育基因,我们在康乃馨基因组中鉴定出同源基因,并构建了两种物种基因的系统发育树。从中,我们在康乃馨中鉴定出35个类型Ⅱ MADS基因,包括五个SEP基因、两个AGL6、三个AP1/FUL、三个SOC1、三个AG/STK、五个AP3/PI(两个PI、两个AP3和一个TM6)、两个Bs、一个AGL12、两个AGL15、三个SVP、四个AGL17和两个MIKC*(图6b)。
为评估康乃馨的复花,将花瓣从外到内分为四轮:外层花瓣、中层花瓣、花瓣状雄蕊和不育雄蕊(图6c)。使用扫描电子显微镜对花瓣的背面进行了表皮变化的评估,从外层到内层。从花瓣到雄蕊的近端表皮细胞形状从不规则圆形变为长圆形,从花瓣到雄蕊的近端表皮细胞从长梭形变为缩短型(图S35)。花瓣状雄蕊的远端背表皮细胞显示了花瓣和雄蕊之间的渐变形状(图S35)。比较了基因表达模式,评估了表达基因的类型和水平,以确定基因表达变化对复花的影响。我们参考了ABC模型对花的发育进行了解读,其中A基因决定萼片,A+B基因决定花瓣,B+C基因决定雄蕊,C基因决定心皮形成(Coen和Meyerowitz,1991;Theißen和Saedler,2001)。我们发现,A类的AP1和FUL在所有组织中
都有表达,但其在花瓣中的表达高于雄蕊,通过qRT-PCR测定。AP2L主要在外层和中层花瓣中表达,而在花瓣状雄蕊中表达水平较低。C类基因的表达不仅在雄蕊中检测到,也在花瓣状雄蕊和中层花瓣中检测到,表达水平在雄蕊中最高,其次是花瓣状雄蕊(图6c)。此外,我们还分析了康乃馨中C类基因的功能(Wang等,2020b)。在我们的结果中,五个B类基因在检测到的所有花器官(外层花瓣、中层花瓣、花瓣状雄蕊和雄蕊)中表达,PI同源基因的表达高于AP3和TM6同源基因(图S36)。
为进一步研究B类基因在花瓣身份中的功能,选择了在B类基因中表达水平较高的DcaPI2进行进一步实验。在两个康乃馨品种‘Lorca’和‘L’中扩增了DcaPI2基因,它们之间存在一个氨基酸的差异(图S37)。我们选择了‘L’中更长的序列‘DcaPI2.2’进行进一步研究。在35S:DcaPI2.2转基因植物中,萼片转变为被分类为花瓣状萼片的器官,基于形状和颜色(图6d)。使用扫描电子显微镜,我们观察到转基因系的萼片边缘细胞专化为花瓣特异性细胞(图S38E)。这些细胞的形状介于野生型拟南芥的萼片表皮细胞(图S38F)和花瓣表皮细胞(图S38G)之间,呈不规则的方形。在拟南芥的转基因系中检测到了DcaPI2.2、AtPI和AtAP3基因的表达,并显示了类似的表达模式(图S39)。这些结果表明PI2在康乃馨中的花瓣识别中起着重要的作用。
在ABC模型的背景下,我们的结果表明,在康乃馨复花形成模型中,A/C类基因表达的边界(A基因向内滑动,C基因向外滑动)模糊,并逐渐淡化,导致花瓣和雄蕊之间的边界融合,从外部(最像花瓣)到内部(最像雄蕊)的花瓣形态逐渐过渡(图6e)。
讨论
在本研究中,我们描述了康乃馨的第一个染色体级基因组装。这一高质量的参考基因组对于康乃馨品种开发者以及研究花的发育和进化的分子机制的研究者来说,是一项重要资源。
康乃馨是世界上最受欢迎的观赏花卉之一,部分原因是其花色、香气和表型的多样性,使其对消费者具有吸引力。以前的研究已经报道了康乃馨色素途径中相关酶和转录因子的功能(Abe等,2008;Fukui等,2003;Momose等,2013;Morimoto等,2019;Yagi等,2013)。然而,这些研究集中在不同品种中代谢物的积累上,并缺乏跨发育阶段的互补基因表达数据,这对于完全阐明潜在途径是必需的。在这里,使用新组装的基因组,结合跨发育阶段和不同组织的转录组和代谢组数据,我们提出了花瓣着色的共表达网络,并推断了关键转录因子的贡献。我们的研究表明,bHLH和MYB一起调节花青素色素的形成,这与其他物种的发现相似(Huang等,2018;Wang等,2021;Xu等,2015)。
近年来,更多的研究发现了WRKY基于调节机制在花青素途径中的保守性,例如在拟南芥中,WRKY因子与MYB-bHLH-WD40(MBW)复合体结合以调节花青素途径(Verweij等,2016)。另一项研究还报道了WRKY蛋白通过与MBW复合体的关系控制类黄酮色素途径(Lloyd等,2017)。此外,WRKY在最近发表的关于杜鹃花颜色的研究中也被发现参与其中(Yang等,2020)。这些研究表明了WRKY因子在色素途径中的重要作用。有趣的是,我们的结果也显示,WRKY44转录因子特异性表达在花瓣边缘,并且预测与DFR基因相互作用,表明它参与康乃馨花瓣边缘的着色。WRKY转录因子在多种不同物种中参与合成花青素;因此,需要更多研究来理解WRKY在被子植物中的进化以及它在其他谱系中的功能。之前在康乃馨中创造新的蓝色色素途径的遗传工程尝试已经成功(Matsuba等,2010),但由于缺乏完整的基因组资源,多色花瓣的努力进展缓慢。众所周知,完整的高质量基因组和基因调控网络的解析可以提高基因编辑技术的效率并减少非目标效应(Manghwar等,2020;Scheben等,2017)。因此,这里提出的高质量基因组和基因调控网络为开发具有新颖多色化的康乃馨品种提供了蓝图,既可以通过常规的标记辅助育种,也可以通过各种基因编辑技术。
为了阐明康乃馨香气的多样性,Clery等(1999)研究了康乃馨的挥发性有机化合物,发现现代品种已经失去了丁香酚的辛辣香气。栽培康乃馨的香气多样性远低于野生石竹属物种。在一项关于花香的回顾性研究中,该研究建议在现代花卉品种中,香气往往无意中被其他性状所淘汰(Schade等,2001;Vainstein等,2001)。在对25个康乃馨品种排放的挥发物进行研究中,发现许多常见栽培品种也失去了产生香气的能力(Kishimoto,2020)。在这项研究中,我们鉴定了参与丁香酚合成的候选基因CFAT和EGS。EGS基因的结构变化可能解释了像‘Francesco’这样的品种缺乏丁香酚的原因。这一发现为测试缺乏香气的品种中这些结构基因差异及潜在的功能基因恢复提供了基础,从而可以再次培育出在保留重要形态和颜色特征的同时具有香气的康乃馨。最后,我们提出,与B类基因一起,A类基因在花轮中的表达可能有助于所有花轮的花瓣形成,并进而成为产生康乃馨中最受追捧的复花形态的一部分(图6d)。总体而言,本研究报告的高质量康乃馨基因组为康乃馨的观赏性状提供了新见解,并为进一步的研究问题和植物生物技术技术的发展提供了基础。
方法
植物材料和组织采集
康乃馨(D. caryophyllus L.)品种‘Scarlet Queen’和‘Lorca’(以花瓣边缘着色著称的知名品种)在中国湖北省武汉市华中农业大学实验生长设施的温室中种植。从‘Scarlet Queen’采集幼叶,用于提取用于Illumina和ONT测序的高质量DNA。从‘Scarlet Queen’的茎、叶和根中获得的RNA-seq数据,以及来自NCBI序列阅读存档数据库(BioProject: PRJDB5916)的已发布数据用于基因组注释。
DNA提取、文库构建和基因组测序
为ONT文库准备,使用Qiagen基因组DNA提取试剂盒(货号10,243和19,060,Qiagen,德国希尔登)按照制造商的说明提取高质量基因组DNA。使用NanoDrop One UV–Vis分光光度计(Thermo Fisher Scientific,美国)进行提取DNA的质量控制,以评估DNA纯度(OD260/280范围从1.8到2.0,OD260/230在2.0到2.2之间)。按照Nanopore文库构建协议,为PromethION测序平台(Oxford Nanopore Technologies,英国)构建文库。对于Illumina测序,我们在HiSeq 2000平台(Illumina,美国圣地亚哥)上产生了约50×Illumina短读,插入大小为400 bp,用以优化ONT基因组组装。使用FastQ (v0.20.0)(Chen等,2018)和FastQC(Brown等,2017)处理原始测序数据,以过滤和检测质量差的读取和过多表达的序列。使用标准程序进行染色质提取和消化,构建Hi-C文库(PE 150bp)。Hi-C文库的详细程序,包括DNA连接、纯化和片段化,在补充说明中描述。Hi-C文库在Illumina NovaSeq平台上测序,所得数据用于构建染色体级基因组组装。
基因组组装和伪染色体支架
使用kmerfreq分析Illumina清洁读取,生成k-mer深度分布,k-mer大小为17 bp。然后使用这些数据估计基因组大小。使用NextDenovo进行ONT长读取的纠正和de novo组装(reads_cutoff: 1k 和 seed_cutoff: 32k)。对于基因组优化,ONT读取和Illumina测序读取分别进行了三轮和四轮基因组校正,使用NextPolish v1.2.4进行(GitHub - Nextomics/NextPolish: Fast and accurately polish the genome generated by long reads.)。使用Purge_Haplotigs子程序(Roach等,2018)生成最终的contig组装,只保留来自杂合区域的每个contig的一个副本。对于Hi-C序列数据,我们还使用fastp初步过滤了低质量读取,并使用Bowtie2 v2.3.2(Langmead和Salzberg,2012)验证配对末端读取。然后我们使用LACHESIS软件(Burton等,2013)(cluster_min_re_sites = 100; cluster_max_link_density = 2.5; cluster_noninformative_ratio = 1.4; order_min_n_res_in_trunk = 60; order_min_n_res_in_shreds = 60)对contig级基因组组装进行聚类、重排和定向,并手动检查Hi-C热图中显现的染色体放置和方向错误。为评估新组装的基因组,使用BUSCO(Simao等,2015)针对Embryophyta_odb10数据库评估基因组组装的完整性。使用Yagi等(2017)发布的‘72L’康乃馨遗传图和ALLMAPS(Tang等,2015)评估质量。最后,通过将我们的Illumina和ONT读取映射回基因组来评判组装质量。
重复元素和基因的注释
在基因预测之前,通过广泛的de novo TE注释器管道(Ou等,2019)掩蔽重复序列。使用基于转录组的、基于同源性的和de novo方法预测高质量蛋白编码基因(补充说明)。对于基于同源性的预测,我们选择了八个物种的转录蛋白序列,包括拟南芥(A. thaliana)、水稻(Oryza sativa)、月季(Rosa chinensis)、葡萄(V. vinifera)、番木瓜(Carica papaya)、康乃馨_draft_r1.0、番茄(Solanum lycopersicum)和甜菜(B. vulgaris)。对于ab initio注释,使用Augustus(Stanke等,2004)、SNAP(GitHub - KorfLab/SNAP: Gene prediction software)和GlimmerHMM(Majoros等,2004)。对于基于转录组的预测,转录组数据是通过对‘Scarlet Queen’的叶、芽和根以及NCBI的花的RNA-seq数据进行Illumina测序产生的。使用HISAT2(Kim等,2015)生成RNA-seq比对文件,并使用PASA程序对剪接转录本进行比对和注释候选基因。使用Evidence Modeler v1.1.1(Haas等,2008)生成使用三种方法获得的基因模型的最终共识集,并使用PASA更新结果。使用BLASTp(e-value 1e-5 cutoff)对蛋白编码基因进行功能注释,使用SwissProt、NR和KOG数据库。使用InterProScan(Jones等,2014)通过搜索InterPro数据库注释蛋白域。从相应的InterPro条目获得每个基因的GO项。使用Rfam和Infernal软件识别非编码RNA。
系统发育分析
为了探索康乃馨 (D. caryophyllus) 的进化历史,我们选择了八个具有完整基因组的物种进行研究,包括甜菜 (B. vulgaris)、甘蓝 (S. oleracea)、荒漠菜 (S. aralocaspica)、胡萝卜 (Daucus carota)、向日葵 (Helianthus annuus)、中国兔 (Nesolagus sinensis)、杜鹃花 (Rhododendron simsii) 和莲花 (Nelumbo nucifera)。我们使用 OrthoFinder (Emms 和 Kelly, 2015) 工具和默认参数生成了用于系统发育分析的矩阵。从这个数据集中鉴定了单拷贝直系同源基因,并用它们构建了系统发育树。蛋白质序列使用 MAFFT (Katoh 和 Standley, 2013) 对齐。我们进一步使用 PAML 包中的 MCMCTree (Yang, 2007) 估计了物种间的分化时间。在估计分化时间时,我们使用了 TimeTree 数据库提供的莲花和向日葵 (121.6–134.9 MYA)、甜菜和荒漠菜 (40–62 MYA)、甜菜和甘蓝 (22–61 MYA)、胡萝卜和向日葵 (77.3–91.7 MYA)、以及向日葵和杜鹃花 (99–114 MYA) 之间的分化时间进行模型校准,并参考了 Cornales 或 Ericales 的顶级年龄为 89.8 MYA(约 90 MYA)(Li et al., 2019)。
通过 Computational Analysis of Gene Family Evolution (Han et al., 2013) 工具,我们鉴定了在八个已测序物种中扩张或收缩的基因家族。通过 BLASTp 全面搜索,我们识别了康乃馨蛋白的同源对,设置 e-value 截断值为 1e−5。使用 MCScanX (Wang et al., 2012) 和默认参数识别了至少包含 15 对同源基因对的共线性块。为了寻找多倍体事件,我们使用 MCScanX 脚本中的 add_ka_and_ks_to_collinearity.pl 计算每对康乃馨_v1、康乃馨 r1_draft、甜菜、甘蓝和莴苣中共线基因的 Ks 值。这些物种的 4DTv 值通过 PAML 包得到。
RNA 测序、基因表达分析和共表达分析
我们收集了 ‘Lorca’ 在不同花发育阶段的样本(如 S1: 活跃的腋芽、茎顶端分生组织转化为花原基;S2: 花分生组织和早期花器官发育;S3: 完全闭合的花;S4: 花色开始出现;S5: 开花前的花蕾和颜色;S6: 开放的花;S7: 花朵衰老),以及红边组织和黄色边缘组织,进行 RNA-seq。我们使用 HISAT2 (Kim et al., 2015) 工具将来自 27 种不同组织的 RNA-seq 短读与我们的染色体级基因组对齐。使用 HTSeq 计算这些样本的读数,并使用 R 包 DESeq2 进行差异基因表达分析(|log2FC| >1 和 padj <0.05
)。我们还收集了不同花器官的样本(外花瓣、中间花瓣、花药状花瓣和花药)进行 qRT-PCR。我们遵循之前文章 (Zhang et al., 2018) 描述的 qRT-PCR 程序和分析方法来量化不同花轮中的基因表达。为了识别差异表达基因之间的关系,我们使用 WGCNA 包 (Langfelder and Horvath, 2008) 构建共表达网络。构建 WGCNA 时使用的参数包括:权重网络、无向、层次聚类树、动态混合树剪切算法、指数 8 和最小模块大小 = 30。然后我们使用 Cytoscape 显示网络。网络统计数据通过 Cytoscape 中的 NetworkAnalyzer (Shannon et al., 2003) 计算。表达热图使用 TBtools 软件 (Chen et al., 2020) 以 log 和行标度生成,并使用在线热图软件 v0.2.2 (Hiplot) 以默认参数生成。
代谢物分析
我们在 S3、S4 和 S6 阶段采集了 ‘Lorca’ 的花瓣并立即在液氮中冷冻。使用 UPLC–ESI–MS/MS 系统 (HPLC, UFLC Shimadzu Nexera X2, www.shimadzu.com.cn/; MS, Applied Biosystems 4500 Q Trap, www.appliedbiosystems.com.cn/) 和 UPLC–APCI–MS/MS 系统 (UPLC, ExionLC™ AD, https://sciex.com.cn/; MS, Applied Biosystems 6500 Triple Quadrupole, SCIEX分析仪器_上海SCIEX爱博才思分析仪器贸易有限公司官方网站) 识别和定量了花青素、类胡萝卜素和黄酮化合物。每个发育阶段评估三个生物学重复的样本。冻干样品按照补充说明中详细的方法进行提取、识别和定量。
‘Lorca’ 花朵释放的挥发性化合物通过固相微萃取收集。这些样品被加入 15 mL 的顶空瓶中,称重并平衡。萃取后,萃取纤维立即插入 GC 注射口进行热分析。每个阶段每个样品进行三个独立的重复。使用 GCMS-QP2010 系统 (Shimadzu, Japan) 进行分析。离子源和界面温度分别为 200℃ 和 250℃。获取花朵化合物的色谱图和质谱图后,通过 NIST2011 库进行定性分析,并根据 Pherobase 数据库中发布的植物挥发性物质进行复核。
植物转化
从 ‘Lorca’ 和 D. chinensis ‘L’ 的花 cDNA 中扩增了 PI2 基因的全长编码序列,使用特异性引物克隆到 pRI101 向量中。使用的引物是 Zhang et al. (2018) 和 Wang et al. (2020) 发表的。所有重组向量通过测序确认。对于遗传转化,我们使用了阿拉伯芥 (Columbia) 植物,采用花浸法 (Logemann et al., 2006) 进行转化。RNA 提取后,使用 SYBR® Premix Ex TaqTM II (Takara, Japan) 在 Applied Biosystems 实时 PCR 系统 (Life Technologies) 中进行 qRT-PCR,每个基因分析三个生物学重复和三个技术重复。相对表达值使用比较性 2−▵▵CT 方法计算。

















 613
613

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









