






总是有小伙伴在学我们的单细胞代码教程(少走点单细胞的弯路)时深感头疼,密密麻麻的代码、繁琐的流程、巨大的数据量,这些都是劝退大家的门槛。这对这一现象,Biomamba联合寻因生物发布了科研利器——SeekSoul Online单细胞云平台及对应的《2周教你学会单细胞生信全套分析》课程,准备好零代码发单细胞文章吧!
课程链接:点击跳转
云平台访问链接:https://seeksoul.online/t_static/Biomamba/Biomamba.html
注册页面:

云平台使用视频教程可参考:单细胞云平台教程全收录
体验反馈问卷链接:https://www.wjx.cn/vm/QIX0xRI.aspx#
各位同学需要联系您的专属在线客服,获取demo数据后,进行操作学习。(点击此处跳转)
图文教程
一、资源总览
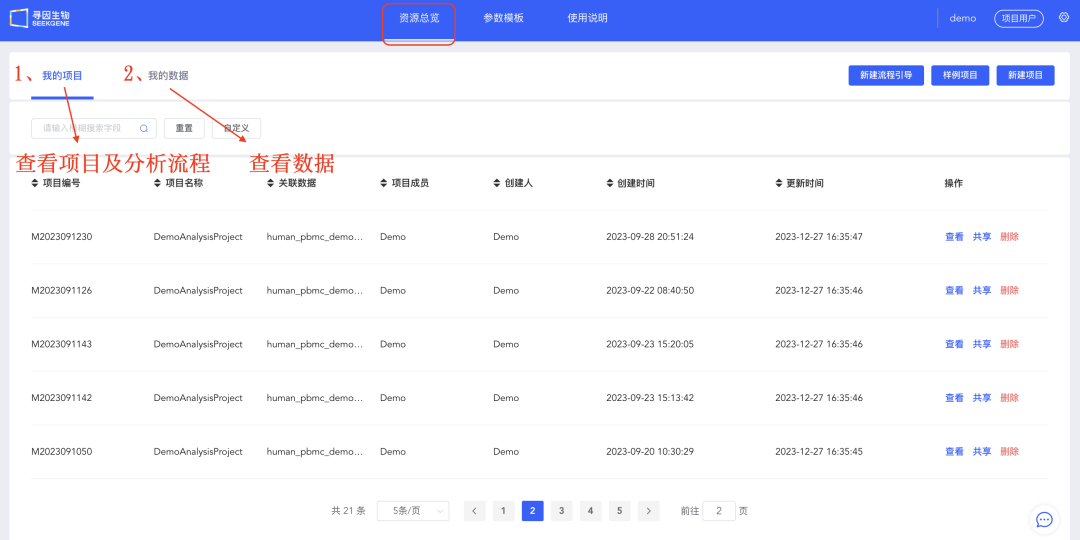
登录云平台后会进入「资源总览」,该界面主要包含四部分内容:
1、《我的项目》:创建项目及单细胞分析流程,可进行「细胞注释」及单细胞相关图片绘制。
2、《我的数据》:样本信息,包括标准分析及用户上传的样本信息。
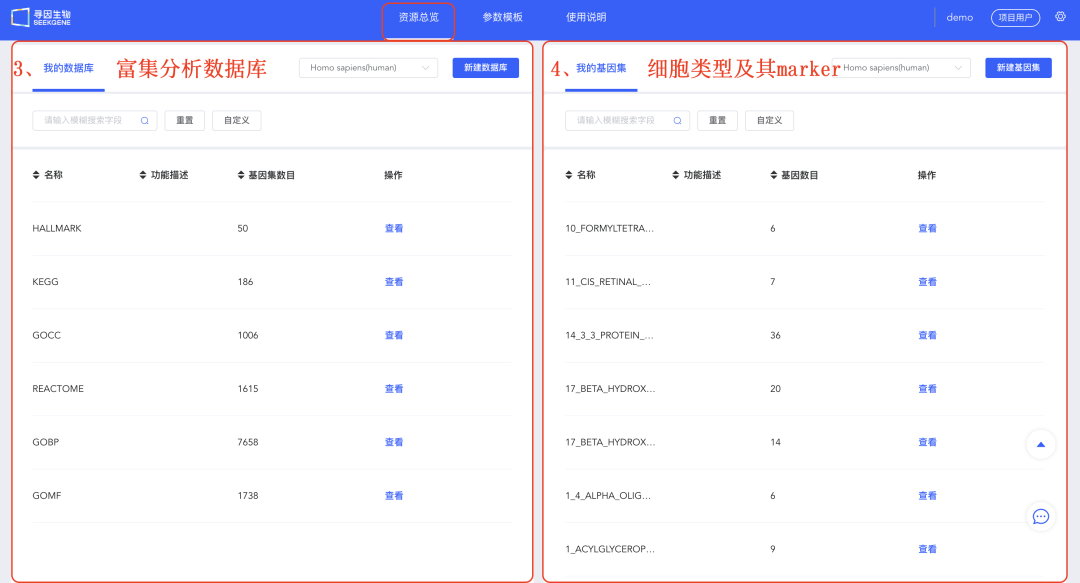
3、《我的数据库》:查看已收集的公共数据库,用户也可以自定义数据库用于后续「差异富集」分析。
4、《我的基因集》:查看已收集的注释细胞类型及其marker,用户也可以自定义marker基因集用于注释及画图。
二、我的项目
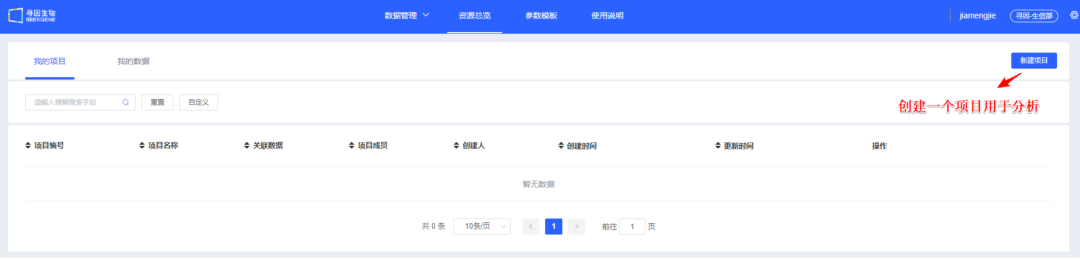
《我的项目》主要用于管理分析流程。同一个项目下可进行大群及亚群的分析,如需新增或删减样本重新分析建议用户创建新的项目。项目创建流程如下:
1、点击【新建项目】。
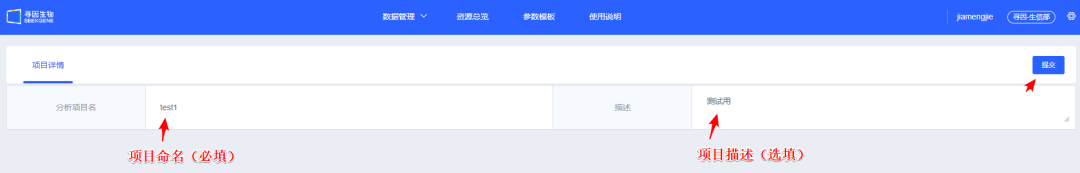
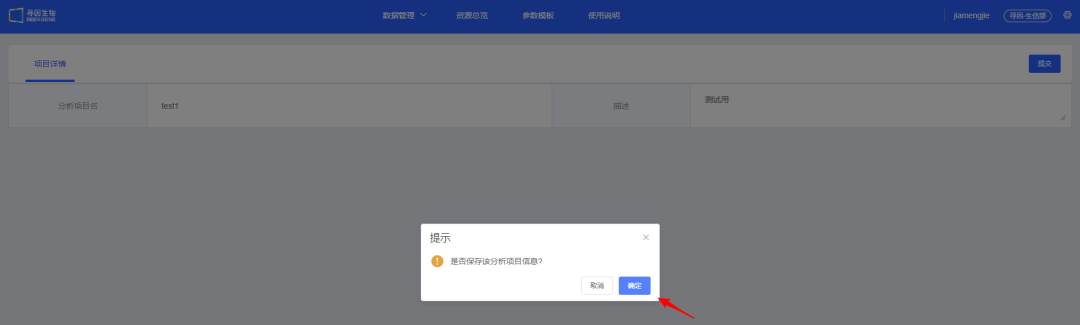
2、填写<分析项目名>及<描述>,【提交】该项目,在弹出的提示信息中点击【确定】保存项目信息。
三、基础分析
「基础分析」是分析流程初始模块,该模块可进行基因线粒体过滤,去批次整合聚类,保留质量合格的细胞进行后续分析。分析流程创建及过滤整合聚类流程如下:
1、【新建流程】创建单细胞分析流程。项目开始一般选择样本进行大群分析,后续选择注释好的细胞类型进行亚群分析。
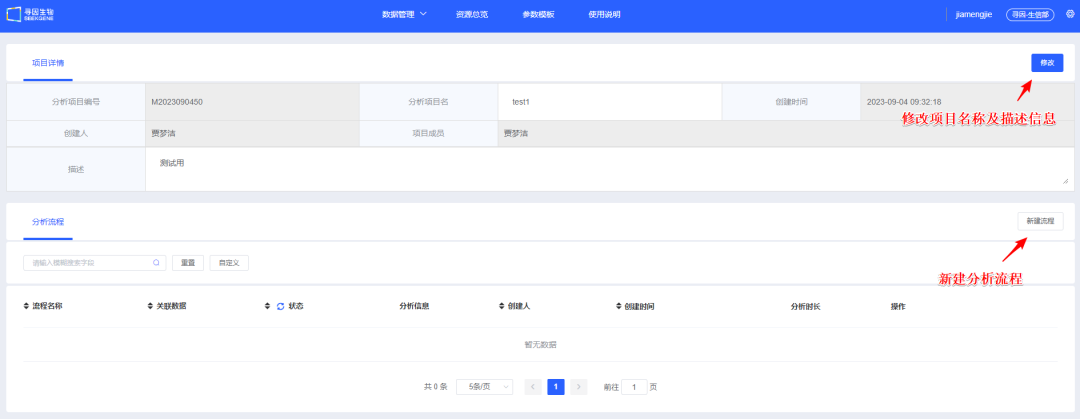
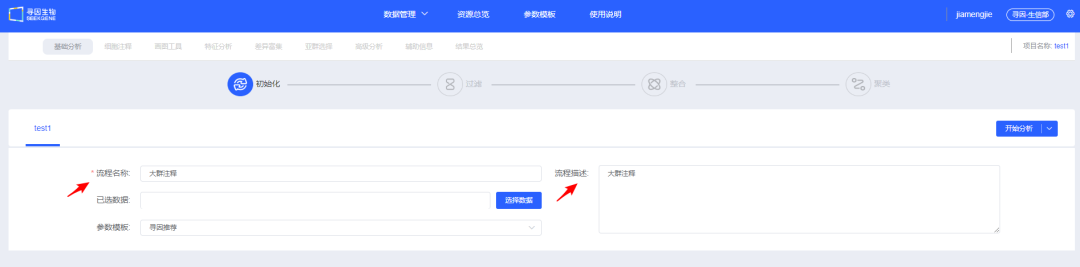
2、填写<流程名称>及<流程描述>,用于后续查找和了解流程信息。
3、【选择数据】选择待分析的样本,可选择已经整合好的多样本数据,也可选多个单样本进行过滤整合。
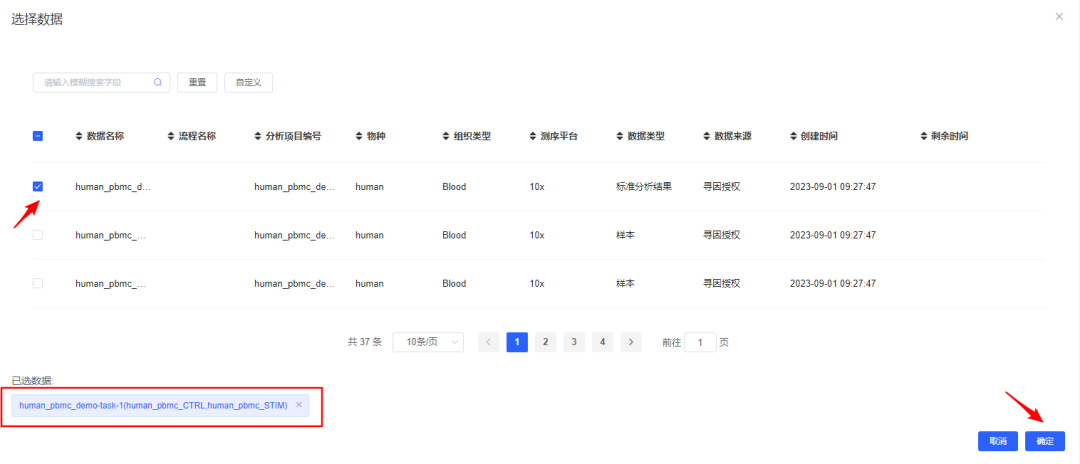
此处数据详细来源可查看《我的数据》。
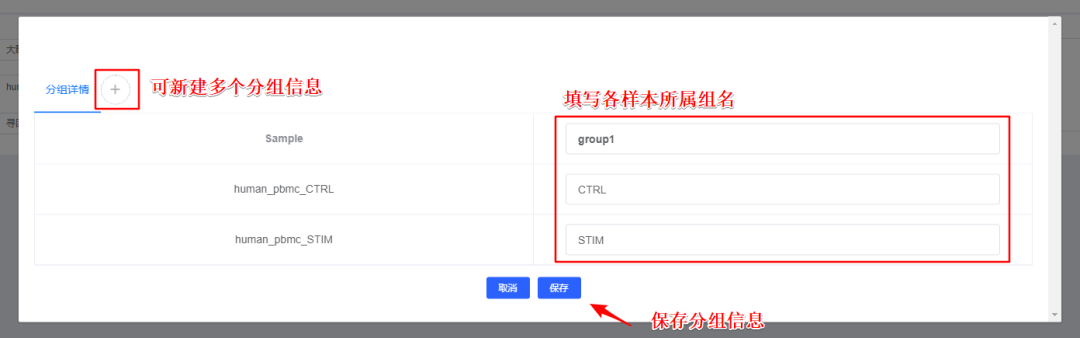
4、【分组】对样本添加分组信息,选填,后续模块也可添加分组信息,详情可见「画图工具」添加标签功能和「差异富集」分组功能。
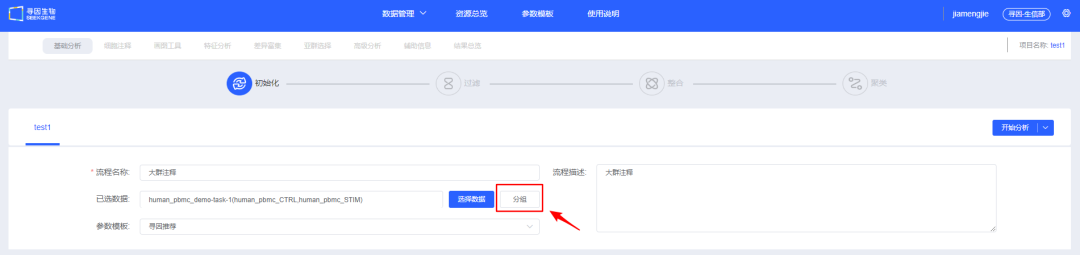
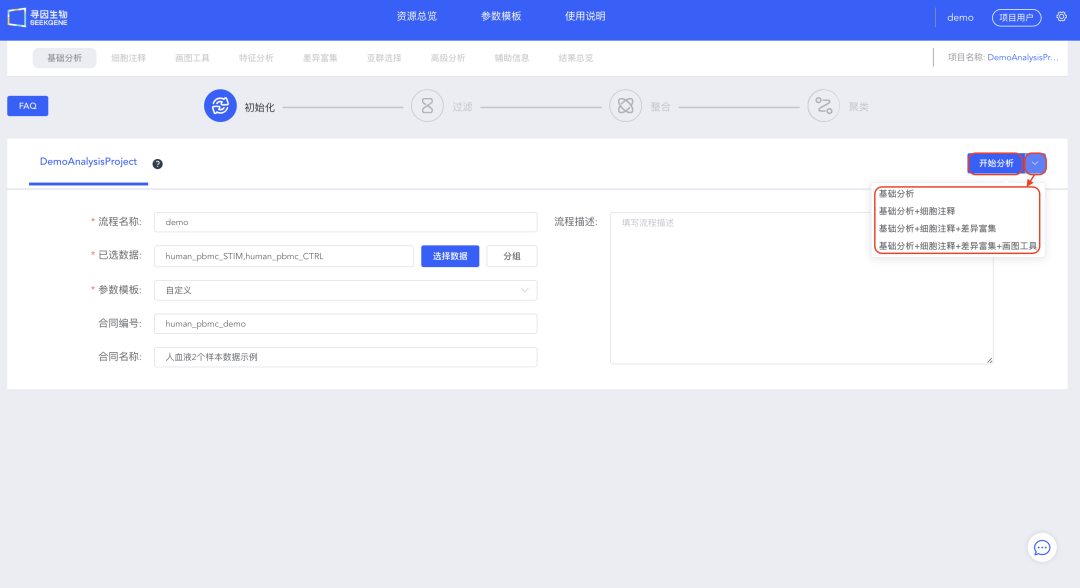
5、填写信息,选择待分析的数据,可以选择【开始分析】手动进行过滤整合和聚类,也可以选择【基础分析】、【基础分析+细胞注释】、【基础分析+细胞注释+差异富集】或【基础分析+细胞注释+差异富集+画图工具】快速进行自动分析。
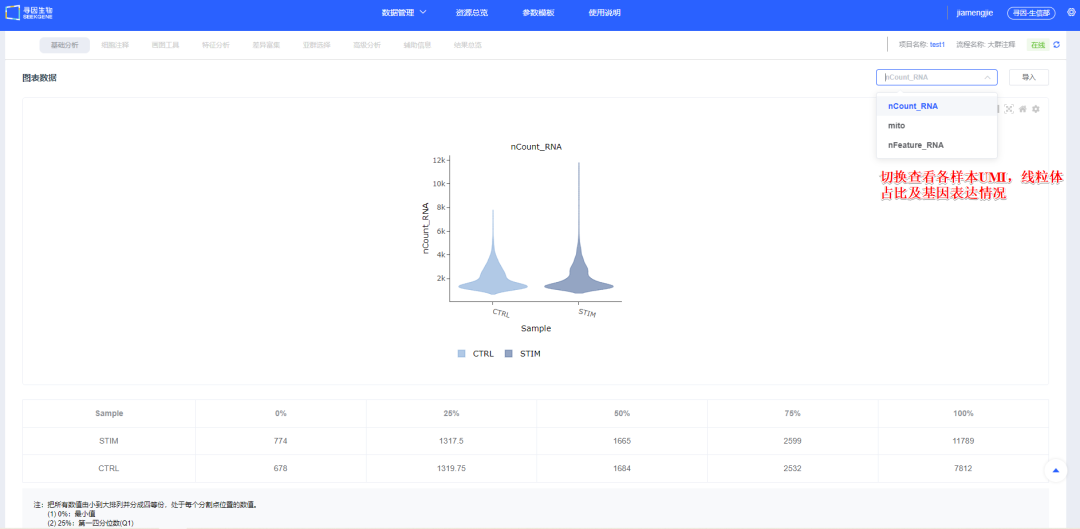
6、【开始分析】后的《图表数据》会统计样本UMI,线粒体占比及基因表达情况,简要展示各样本质量信息。
7、展开按钮查看默认参数,用户可参考已发表单细胞文章方法中的过滤参数进行【过滤】。
注:如果选择的是已经整合的数据,点击【过滤】会提示是否需要跳过过滤整合步骤,如果不需要调整参数可跳过进行后续分析,如果需要调整过滤阈值则取消跳过,重新进行过滤整合。
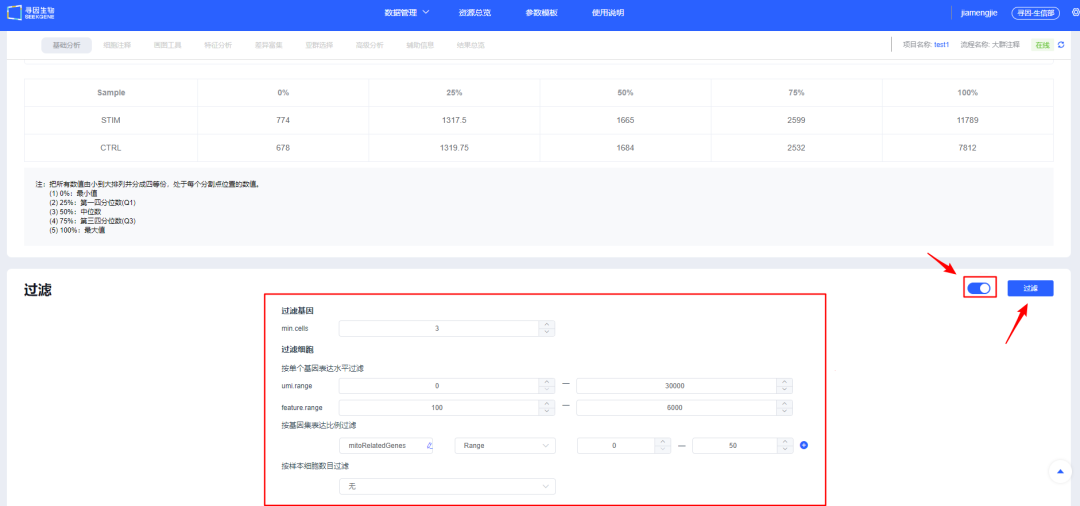
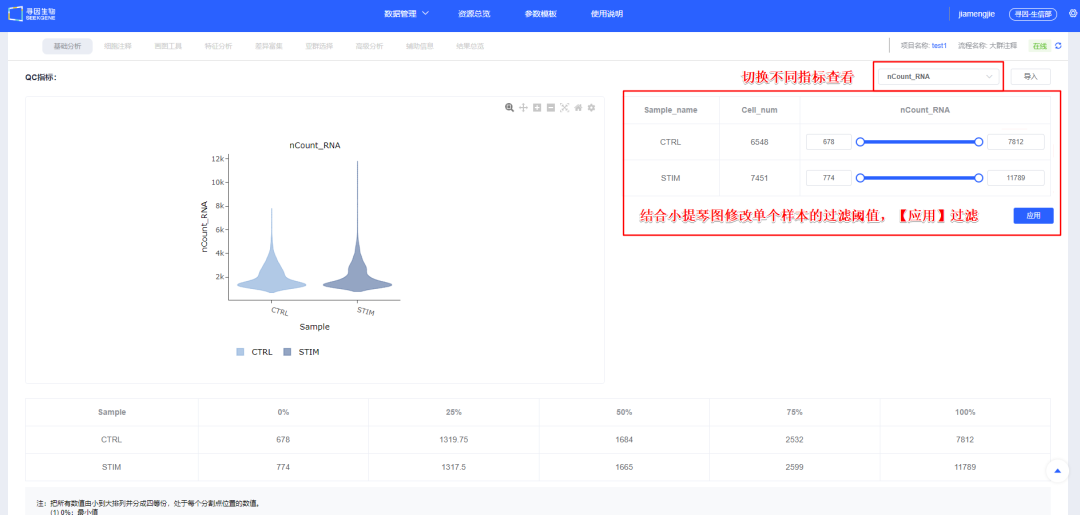
8、【过滤】后可查看各样本过滤后质量信息,同时可对单样本进行个性化调整,保证整体样本质量一致。
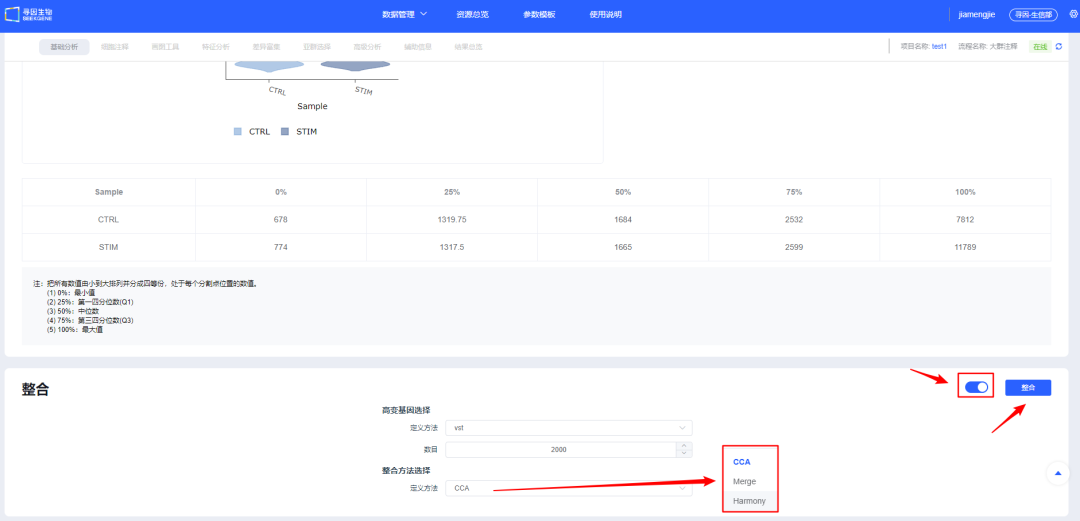
9、质控合格的数据进行【整合】,目前提供三种整合方法,其中 CCA 和 Harmony 会对多个样本进行批次矫正。
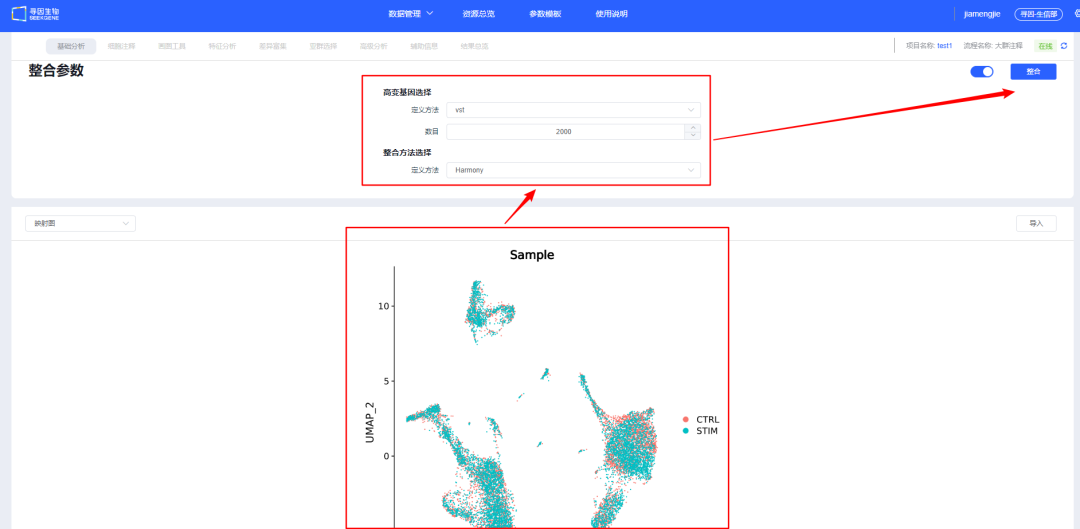
10、【整合】后会展示样本整合情况,可调整整合参数重新整合。建议用户尝试调整多种整合方法,选择更合适的方法进行后续分析。
11、【整合】确认后进行【聚类】,可新建选择多个分辨率进行聚类,分辨率越大分群数量越多。后续模块也可新增分辨率进行聚类。
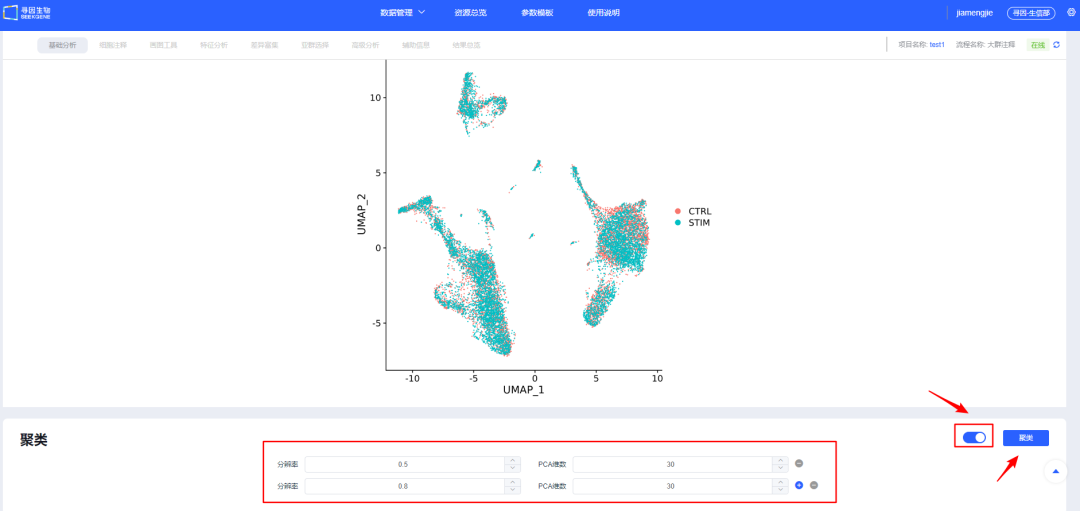
12、【聚类】后无需调整则点击【完成】,跳转「细胞注释」模块正式开始进行单细胞相关分析。该步会耗费一些时间,请用户耐心等待。
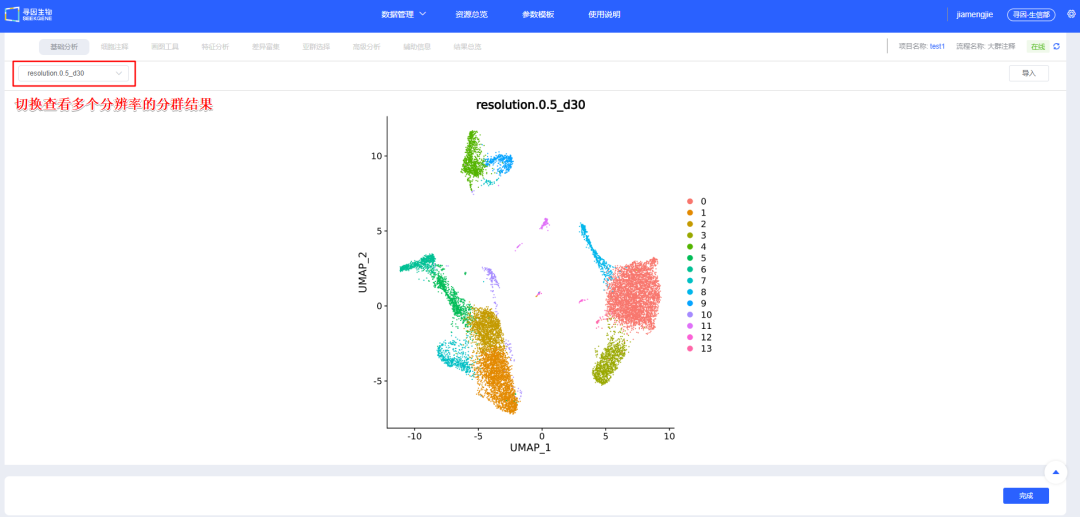
四、细胞注释
完成所构建项目的「基础分析」后,可进入「细胞注释」模块,该模块展示了细胞注释前后的降维图、cluster差异基因列表及top基因的可视化结果,并且可分别对多组聚类结果进行自动注释(singleR算法)和手动注释。
1、主要内容:
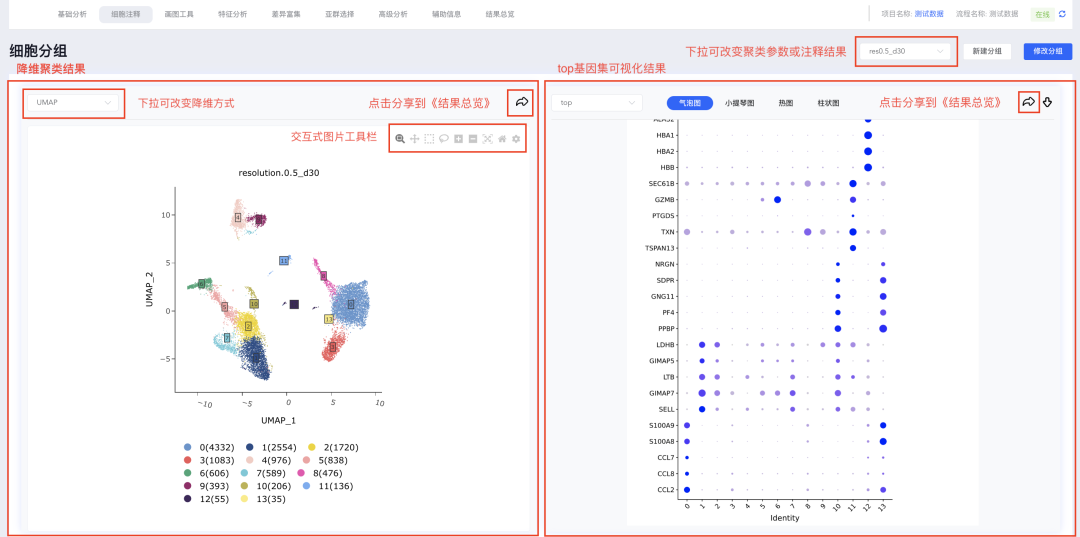
「细胞注释」 模块主要包括四部分内容:降维聚类结果、top基因或指定基因集的可视化结果、cluster差异基因列表及差异基因的表达情况。点击选择降维聚类结果,界面中相关结果也会随之发生改变。
(1)降维聚类结果包括UMAP和tSNE两种降维方式的结果,降维聚类图为交互式图片,可使用鼠标更改cluster的标签位置和查看cluster的细胞数。
(2)每次更新聚类或者注释结果时,均会计算每个cluster的差异基因列表,并且对每个cluster的top 5(logFC从大到小排序)基因进行气泡图、小提琴图和热图可视化展示以及每个样本中每组细胞的占比柱状图。
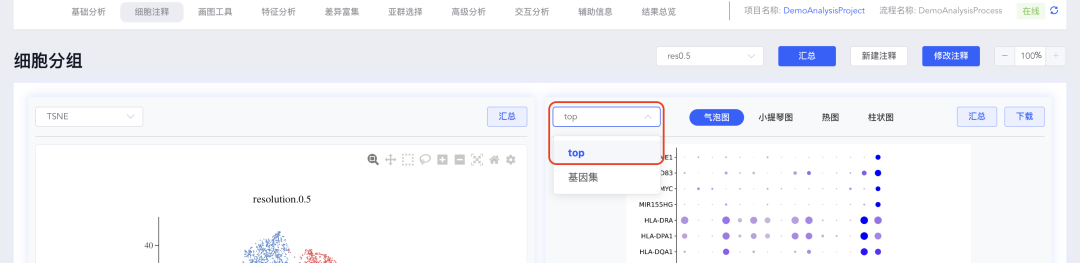
(3)top基因集可视化结果可以自定义修改展示基因,点击【top】弹出每组的基因列表,勾选或不勾选对应的基因后,点击【确定】即可对所选的基因进行重新绘图;同时也可以对指定基因进行可视化展示,点击top下拉中的【基因集】弹出基因集窗口,可从寻因数据库中选择相关基因集,也可以点击分析流程基因集后面的【+】自定义基因集进行可视化展示。
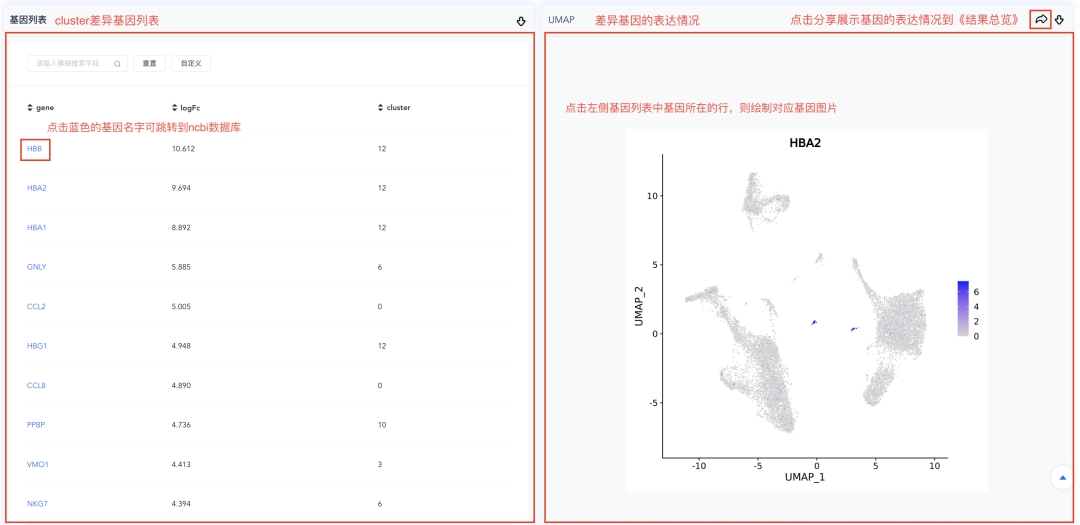
(4)基因列表记录所有基因在某个cluster或分组相对于其他细胞的logFC值。点击基因名称跳转到NCBI网站,点击基因所在的行,可绘制该基因的FeaturePlot图(位于列表右侧)。该列表默认按照logFC从大到小排序,点击表头可自定义排列顺序。
2、自动注释:
「细胞注释」模块主要可进行的分析包括自动注释和手动注释,在完成「基础分析」后,会基于最小分辨率,使用singleR软件及其参考集(人:HumanPrimaryCellAtlasData_main,小鼠和大鼠:ref_Mouse_all)进行自动注释,自动注释的标签为CellAnnotation。
(1)首先下拉选择自动注释要使用的分辨率,然后点击【新建分组】,出现添加分组弹窗,该弹窗可随意拖动位置(鼠标放置在可拖拽区域)和大小(鼠标放置在弹窗右下角)。
(2)填写<分组名称>,并选择<自动注释参考集>,最后点击【自动注释】,自动刷新分析进度。
(3)分析进度完成后,对应的注释结果自动填充到分组详情表中。同时,降维图、top基因可视化结果、基因列表和基因可视化图同步更新。注释完成后后点击弹窗底部的【关闭】,关闭弹窗。
(4)点击同一分辨率下新增分组名称的注释标签,可进行结果的切换。
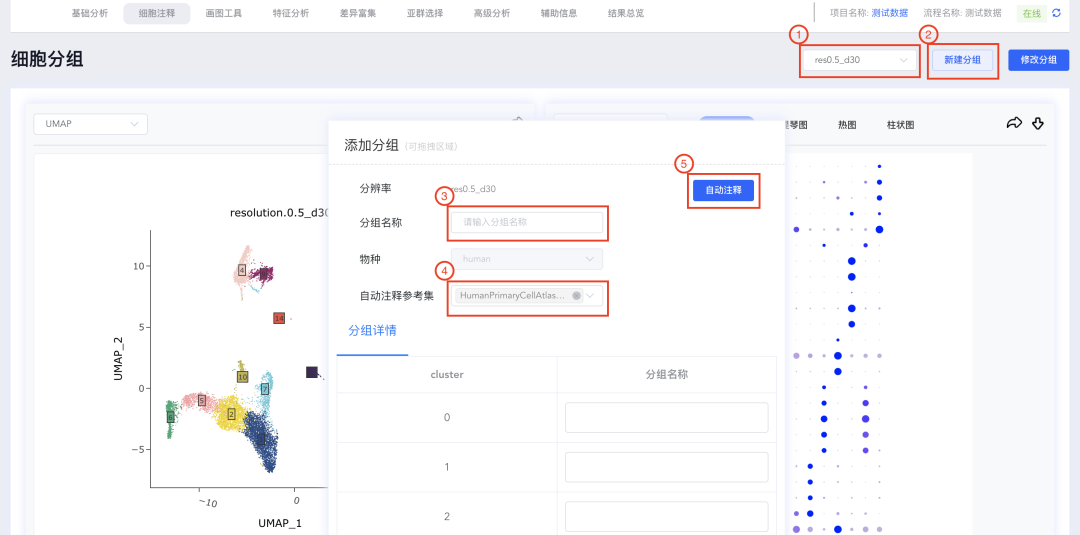
3、手动注释:
当我们依据cluster间的差异基因或收集到的一些marker基因的表达情况,需要输入或修改cluster的注释结果时,可进行如下相关手动注释的操作。
(1)首先下拉选择手动注释要使用的分辨率,然后点击【新建分组】,出现添加分组弹窗,该弹窗可随意拖动位置(鼠标放置在可拖拽区域)和大小(鼠标放置在弹窗右下角)。
(2)填写<分组名称>,在分组详情中输入每个cluster对应的细胞类型。
(3)每个cluster对应的的细胞类型输入完成后,点击弹窗底部的【保存】,出现分析进度条,完成后点击【关闭】按钮,关闭弹窗。
(4)点击同一分辨率下新增分组名称的注释标签,可进行结果的切换。
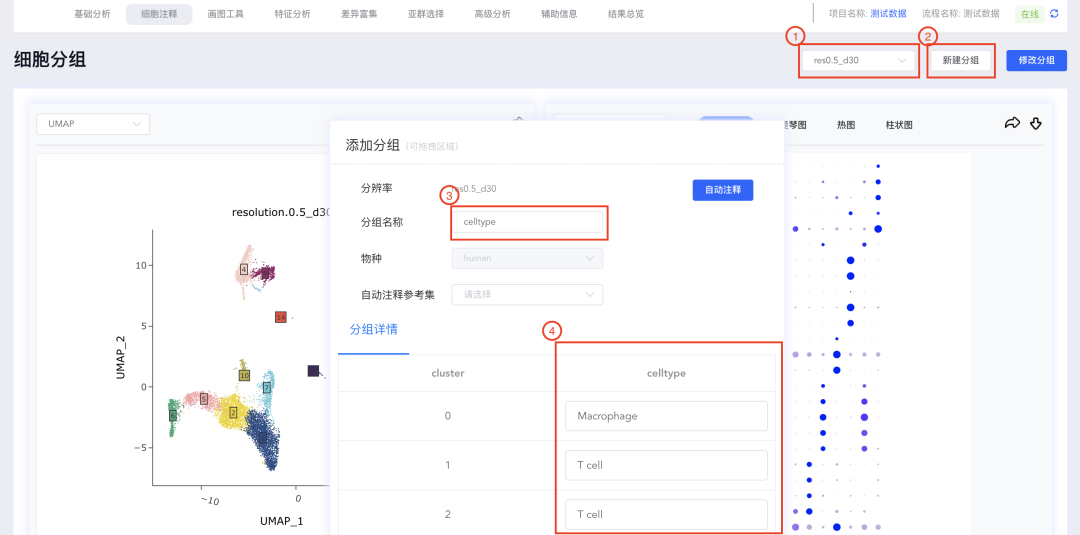
(5)新建分辨率:
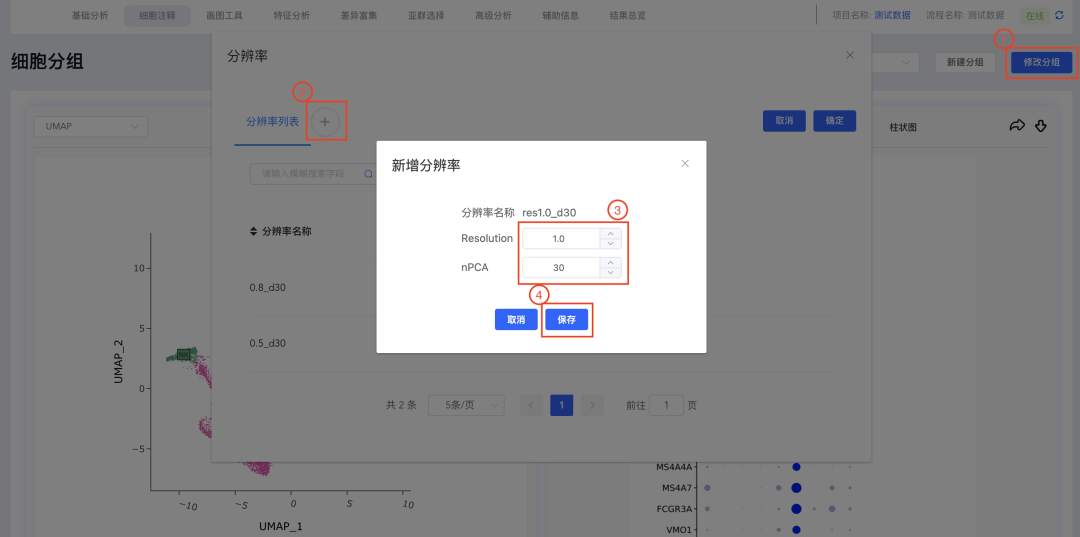
点击细胞分组中的【修改分组】弹出分辨率窗口,点击分辨率列表后面的【+】弹出新增分辨率窗口,输入分辨率(Resolution)和PC数量(nPCA)后,点击【保存】,即可基于新的分辨率和PC数量重新进行聚类分析。完成后,细胞分组下拉框和分辨率列表均新增对应结果,点击细胞分组可进行结果切换。
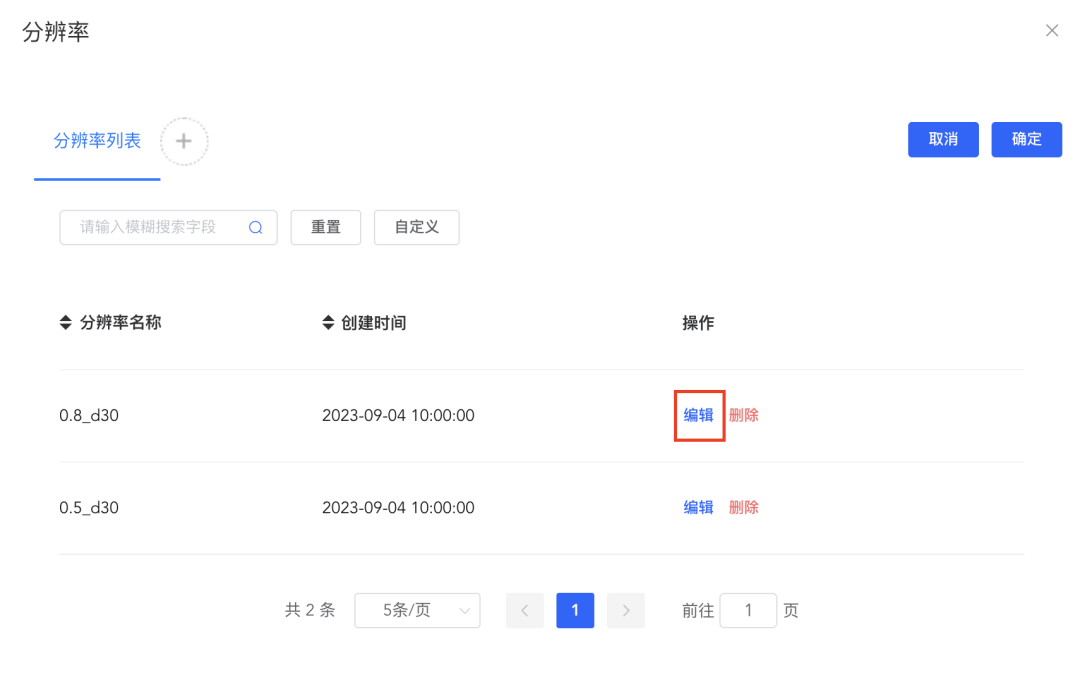
(6)修改注释结果:
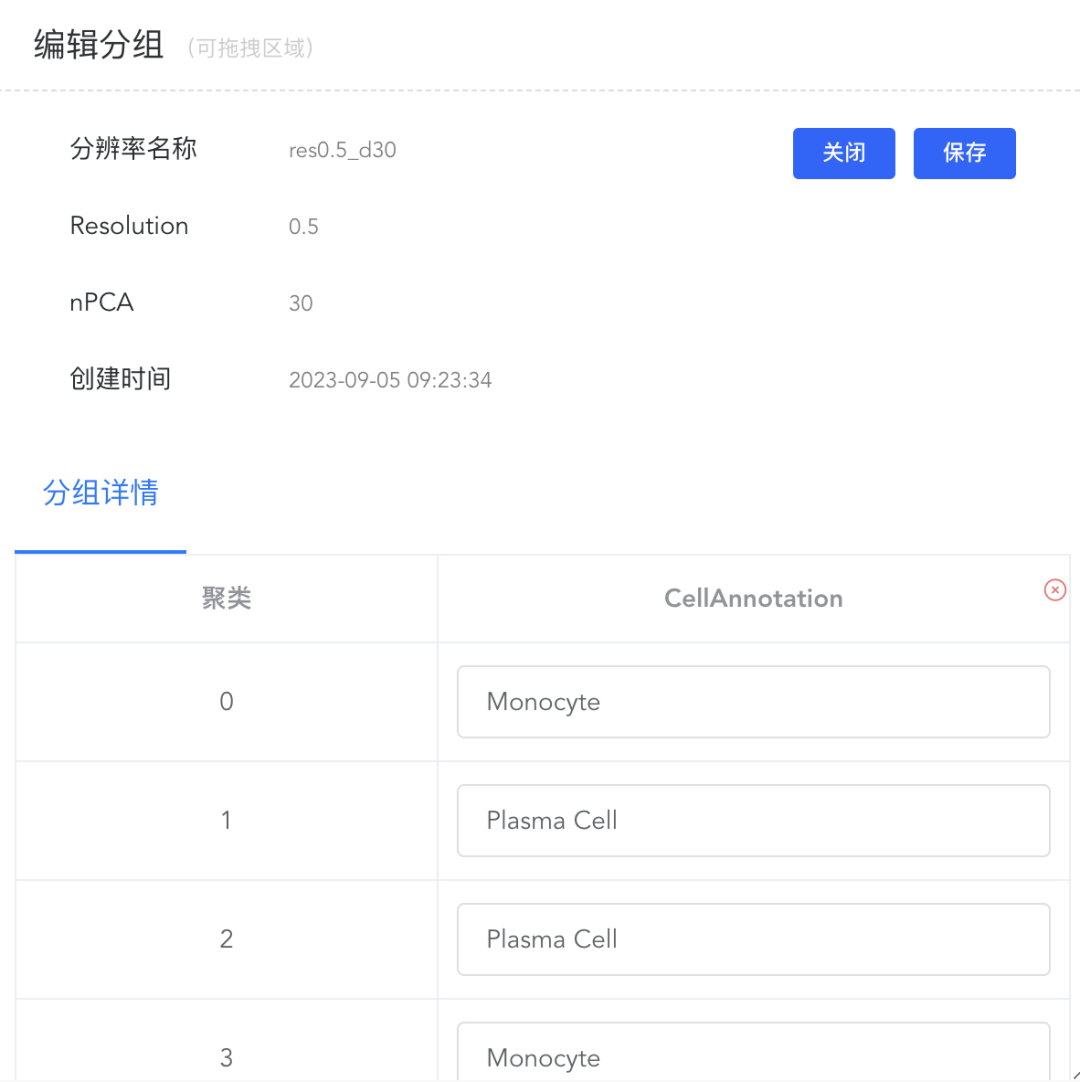
点击细胞分组中的【修改分组】弹出分辨率窗口,点击分辨率对应的【编辑】可修改注释结果,如果该分辨率未做任何注释,则弹出暂无分组的提示信息;点击【删除】即可删除该分辨率及对应的细胞注释结果。
(7)保存注释结果:
点击【编辑】后弹出编辑分组弹窗,分组详情中则包括此分辨率下所有cluster的标注结果,可进行相应的编辑处理,修改完成后点击【保存】同步结果。点击【x】可删除该分辨率下的该组注释结果。
(8)注意事项
a.可以针对同一个降维聚类结果进行多次注释并修改注释;
b.可比较使用不同方法、参考集的注释结果差异;
c.当细胞数量过大时,热图会丢失label等信息,因此热图是经过降低数据量(downsample)绘制的,每个cluster或分组最多保留500个细胞;
d.为了加快计算差异基因速度,该模块基因的logFC是基于edgeR包计算得到的,与FindAllMarkers或者FindMarkers结果会存在些许差异,但两种方法计算出的基因呈线性相关;
e.降维聚类结果、top基因或指定基因集的可视化结果及差异基因的表达情况均可分享到「结果总览」,方便结果的查看和导出。
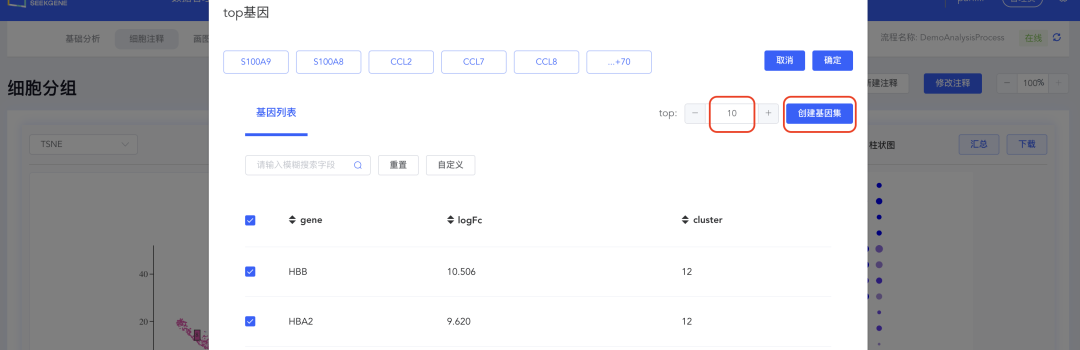
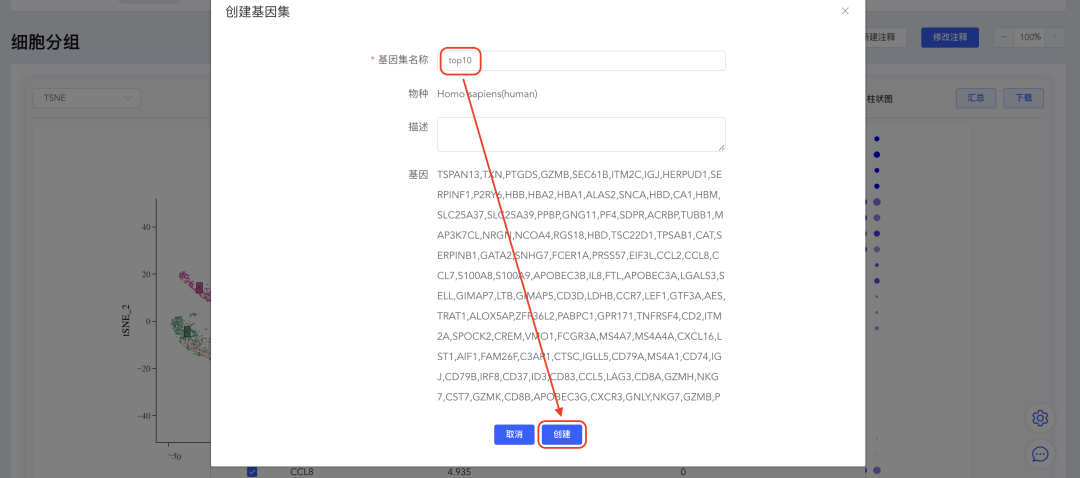
4、创建top N基因集
点击【top】按钮打开top基因界面,填写top基因的数量(如top10),点击【创建基因集】,填写基因集名称,点击【创建】完成基因集的创建。

















 762
762

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









